
ITS2 Sequencing Protocol
Putnam Lab ITS2 Sequencing Protocol
Prerequisites:
- Snap-frozen or well-preserved tissue samples
- DNA extracted from tissue samples (Putnam Lab Zymo Duet RNA DNA Extraction Protocol)
- Quantity and quality of DNA checked (Quality: Gel Electrophoresis and Quantity: Qubit)
Resources:
- PCR and Gel Electrophoresis Descriptions and Troubleshooting: Strand 2017
- Khan Academy: PCR Explained
- Internal Transcribed Spacer 2 (ITS2) is a region of ribosomal DNA: Universal DNA Barcode
- DNA Barcoding Explained: International Barcode of Life, Barcoding 101
Materials:
- Primers: ITS2
| Gene | PrimerName | Sequence (5’-3’) | SequenceWithPartialIlluminaTail (5’-3’) |
|---|---|---|---|
| ITS2 | ITSintfor2 | GAATTGCAGAACTCCGTG | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGAATTGCAGAACTCCGTG |
| ITS2 | ITS2_Reverse | GGGATCCATATGCTTAAGTTCAGCGGGT | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGGGATCCATATGCTTAAGTTCAGCGGGT |
Citation: LaJeunesse TC, Trench RK (2000) Biogeography of two species of Symbiodinium (Freudenthal) inhabiting the intertidal sea anemone Anthopleura elegantissima (Brandt). Biol Bull 199: 126−134.
- Phusion HiFi Mastermix (Thermo Scientific F531S): https://www.fishersci.com/shop/products/phusion-high-fidelity-pcr-master-mixes/f531s
- Ultra-pure water
- 96-well plates, centrifuge, pipettes and filter tips, thermocycler
Protocol:
We start with 10 ng of DNA added to the master mix for this protocol.
- Check concentrations with Qubit.
- Calculate volumes of DNA and water needed to dilute sample to 3.33 ng/μl concentration in 10 μl. 3.33 ng is the minimum input to be used for one reaction. In this protocol, samples will be run in triplicate with each reaction containing 3.33 ng for a sample total of 10 ng. Dilutions can be done in any volume, but we recommend 10 or 20 μl depending on your starting concentration.
| Sample ID | DNA (ng_μl) | DNA for dilution (μl) | Water for Dilution (μl) |
|---|---|---|---|
| Example | Qubit value | =33/Qubit value | 10 - DNA for dilution value |
| Coral 1 | 6.82 | 4.84 | 5.16 |
To calculate the DNA sample volume needed for the dilution in 10µl for example, use the following equation: V1M1=V2M2.
V1(Qubit value)=(3.33 ng/μl)(10 μl)
V1=33/Qubit value
Finally, to calculate the volume of water needed, subtract the DNA volume required from 10 μl.
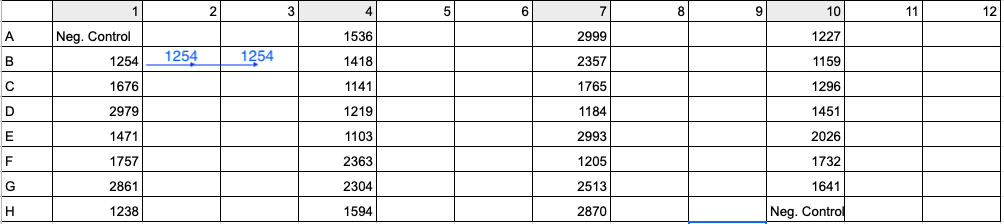
- Aliquot the appropriate volume of ultra pure water needed for dilution (for Coral 1, 5.16 μl) into each appropriately labeled PCR strip tube or well in a 96-well plate. To maximize efficiency, copy the PCR plate setup from Step 11 and the example image. Column 1 in the PCR 96-well plate should be the same as column 1 in the dilution plate so that you can use the multi-channel pipette to add 3 uL in Step 8.
- Aliquot the appropriate volume of DNA sample needed for dilution (for Coral 1, 4.84 μl).
10 ng is widely used as a general starting point, this is usually enough DNA to amplify your desired gene. If the sample is suspected to contain more inhibitors, decrease this starting value. If the sample is not amplifying, a troubleshooting option is to decrease and increase this value. Starting with 10 ng in one 100 μl well that is split into triplicate wells for the PCR steps, 3.33 ng of DNA is needed per reaction (3.33 ng in 33 uL per reaction). Standardize the DNA concentration of each sample prior to amplification.
- Make positive mixture control by adding 0.5 μl of each sample into its own well (if doing dilutions on plates) or tube (if doing dilutions in PCR strip tubes).
- Make master mix stock solution. Forward and reverse primers will come in 100 uM stock solutions, dilute this to 10 uM. Keep master mix stock solution on ice.
| Component | Per Rxn | FINAL CONC | ITS2 |
|---|---|---|---|
| 2X Phusion Mastermix | 50 μl | 1X | 1750 |
| F primer (10uM) | 2 μl | 0.4uM | 70 |
| R primer (10uM) | 2 μl | 0.4uM | 70 |
| H2O | Up to 100 μl (45 μl) | 1645 |
- Add 97 μl of master mix stock solution to each well.
- Add 3 μl of DNA sample (from the 10 μl dilution mix) to each well.
- Add 3 μl ultra pure water to one well per plate as a negative control.
- Add 3 μl of the positive mixture control to one well (total, not per plate) as a positive control.
- Set up each reaction in duplicate or triplicate for the PCR by pipette mixing and moving 33 μl of DNA/master mix solution into each neighboring empty well or PCR strip tube.
See below image for an example of 96-well plate setup with 30 samples (four digit values) and 2 negative controls (Neg. Control). To run samples in triplicate, add DNA and master mix to columns 1, 4, 7, and 10. Then in Step 11 above and image below, move 33 uL of Sample 1254’s reaction from B1 to B2 and 33 uL of reaction from B1 to B3. Use a multi-channel pipette to save time and energy.
- Spin down plate.
- Run the following PCR program (this cycle program is specific to ITS2 for this primer set):
| Cycles | Time | Temp |
|---|---|---|
| 1 | 3 min | 95° |
| 35 | 30 sec | 95° |
| 30 sec | 52° | |
| 30 sec | 72° | |
| 1 | 2 min | 72° |
| 1 | ∞ min | 4° |
- Run 5 μl of each product on a 2% agarose gel using the following Putnam Lab Gel Electrophoresis protocol to check for ~300 bp product.
- Pool products into PCR strip tubes (100 μl total) appropriately labeled with the sample ID. Pooling occurs only after duplicate or triplicate samples (based on if you chose duplicates or triplicates above) have successfully amplified and confirmed on the gel. These will be the PCR product stock. Store at -20°C.
- Aliquot 45 μl of each product from the PCR product stock (in Step 11) into new PCR strip tubes appropriately labeled with the sample ID. These tubes will be delivered to the sequencing center. Store at -20°C until delivering to the sequencing center.
Example of google spreadsheet for data processing, including master mix and dilution calculations, and 96-well PCR platemaps.
Sequencing details from Janet
A two-step PCR protocol was used to prepare PCR products for sequencing according to a modification of Illumina’s 16S Metagenomic Sequencing Library Preparation protocol. A first round of PCR was performed using insert your methods here. Samples were then submitted to the RI-INBRE Molecular Informatics Core at the University of Rhode Island (RI, USA) for the index PCR and sequencing.
In brief, PCR products from the first round of PCR were cleaned with NucleoMag beads (Takara Bio, USA) and then visualized by agarose gel electrophoresis. A second round of PCR (50 ng of template DNA, 8 cycles) was performed to attach Nextera XT indices and adapters using the Illumina Nextera XT® Index Kit (Illumina, San Diego, CA) and Phusion HF PCR mastermix (Thermo Fisher Scientific, US). PCR products from the second PCR were cleaned with NucleoMag beads and analyzed by agarose gel electrophoresis. Selected samples were run on an Agilent BioAnalyzer DNA1000 chip (Santa Clara, CA). Quantification was performed on all samples prior to pooling using a Qubit fluorometer (Invitrogen, Carlsbad, CA, USA), and the final pooled library was quantified by qPCR in a Roche LightCycler480 with the KAPA Biosystems Illumina Kit (KAPA Biosystems, Woburn, MA). Samples were analyzed using 2xyour run bp paired-end sequencing on an Illumina MiSeq (Illumina, San Diego, CA).