
SNP Calling with RNASeq data
SNP Calling with RNASeq Data
Goal: Call SNPs for my Bleaching Pairs project using RNASeq data. I originally tried EpiDiverse to call SNPs with my WGBS data but am continuing to troubleshoot that pipeline (link here).
I recommend looking at the below references in parallel with this script.
References:
- Palumbi lab workshop: https://github.com/UCcongenomics/Conservation-Gene-Expression/blob/master/UCGenomics-SNPcalling-Palumbilab_final%20(1).pdf; https://github.com/bethsheets/SNPcalling_tutorial
- Federica’s pipeline: https://github.com/fscucchia/Pastreoides_development_depth/tree/main/SNPs (Federica referenced Tim’s)
- Tim’s pipeline: https://github.com/TimothyStephens/Kaneohe_Bay_coral_2018_PopGen
Data path = /data/putnamlab/estrand/BleachingPairs_RNASeq/SNP.
All analysis done on URI’s HPC system Andromeda.
Workflow
https://nbisweden.github.io/workshop-ngsintro/2005/lab_vc.html#5_Variant_calling_in_cohort
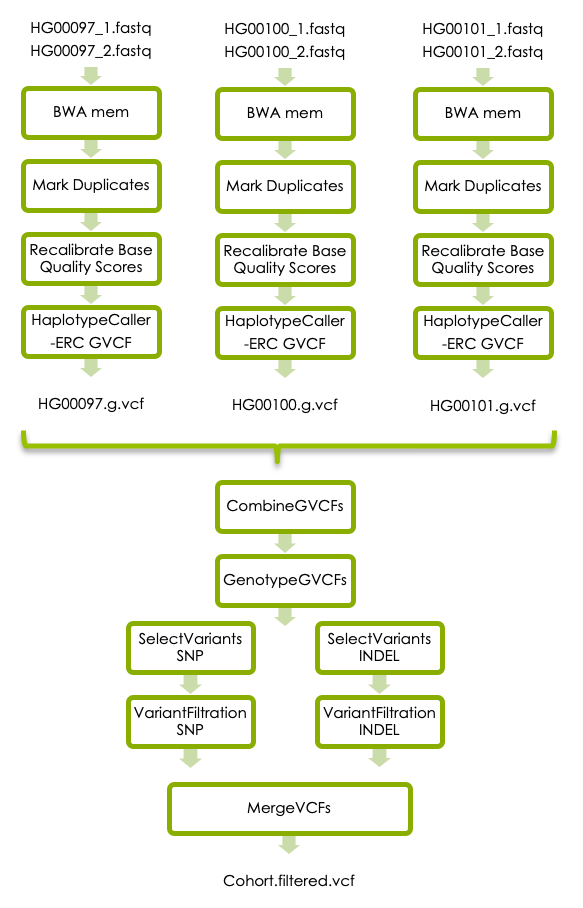
Input
/data/putnamlab/estrand/BleachingPairs_RNASeq/processed/trimmed.*.fastq.gz. Both R1 and R2
0. Installing Programs
- GATK (https://gatk.broadinstitute.org/hc/en-us/articles/360036194592-Getting-started-with-GATK4). Version already downloaded on Andromeda:
GATK/4.3.0.0-GCCcore-11.2.0-Java-11. - rgsam (https://github.com/djhshih/rgsam). I asked Kevin Bryan to download
rgsam/0.2.1-foss-2022a. - PLINK (https://www.cog-genomics.org/plink2). Version already downloaded on Andromeda:
PLINK/2.00a2.3_x86_64.
01. FastqToSam, collect RG, and MergeBamAlignment
1a. FastqToSam
https://gatk.broadinstitute.org/hc/en-us/articles/360037057412-FastqToSam-Picard-.
Purpose: Converts a FASTQ file to an unaligned BAM or SAM file.
Input: One FASTQ file name for single-end or two for pair-end sequencing input data. These files might be in gzip compressed format (when file name is ending with “.gz”).
Output: A single unaligned BAM or SAM file. By default, the records are sorted by query (read) name.
FastqToSam.sh:
Mine took ~7.5 minutes per sample (x40) = 300 minutes.
#!/bin/sh
#SBATCH -t 200:00:00
#SBATCH --export=NONE
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/SNP
#SBATCH --error=output_messages/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=output_messages/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
# Load modules needed
module load GATK/4.3.0.0-GCCcore-11.2.0-Java-11
# Data path to filtered reads
path="/data/putnamlab/estrand/BleachingPairs_RNASeq/processed"
# Create a list for every file that ends with R1__001.fastq.gz in that folder
array1=($(ls $path/*_R1_001.fastq.gz))
# A for loop that says for every file that ends with R1 and R2
for i in ${array1[@]}; do
gatk FastqToSam \
--FASTQ ${i} \
--FASTQ2 $(echo ${i}|sed s/_R1/_R2/) \
--OUTPUT ${i}.FastqToSam.unmapped.bam \
--SAMPLE_NAME ${i}; touch ${i}.FastqToSam.done
done
Tips
The sed argument ‘s/_R1.fastq//’ has 3 parts. Each is separated by /.
- s means substitution
- _R1.fastq is something to be substituted
- The next is actually empty, i.e., substitute _R1.fastq with nothing
1b. collect RG
I moved all the unmapped bam files produced above into a new directory within my SNPs folder.
Purpose: fill in
Input: ${i}.FastqToSam.unmapped.bam from the previous script
Output: ${i}.rg.bam ; edited bam file
collectRG_rgsam.sh: 10-12 hour range.
#!/bin/sh
#SBATCH -t 200:00:00
#SBATCH --export=NONE
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/SNP
#SBATCH --error=output_messages/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=output_messages/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
# Load modules needed
module load SAMtools/1.16.1-GCC-11.3.0
module load rgsam/0.2.1-foss-2022a
# Data path to unmapped bam files reads
path="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP"
# Create a list for every file that ends with FastqToSam.unmapped.bam in that folder
array1=($(ls $path/unmapped_bam/*FastqToSam.unmapped.bam))
# A for loop that says for every file that ends with R1 and R2
for i in ${array1[@]}; do
samtools view ${i} | rgsam collect -s ${i} -o ${i}.rg.txt
samtools view -h ${i} |
rgsam tag -r ${i}.rg.txt |
samtools view -b - > ${i}.rg.bam
done
Tips
https://github.com/djhshih/rgsam
rgsam commands:
- collect: collect read-group information from SAM or FASTQ file
- split: plit SAM or FASTQ file based on read-group
- tag: tag reads in SAM file with read-group field
- qnames: list supported read name formats
- version: print version
Note that we use the -h flag of samtools view to ensure that other header data are preserved (any existing @RG will be replaced)
Troubleshooting
I ran into several errors with versions of modules. rgsam/0.2.1-foss-2022a needs to be pairs with GCC-11.3.0 so Kevin Bryan downloaded SAMtools/1.16.1-GCC-11.3.0 and that seemed to work.
GCCcore/11.3.0(24):ERROR:150: Module 'GCCcore/11.3.0' conflicts with the currently loaded module(s) 'GCCcore/11.2.0'
GCCcore/11.3.0(24):ERROR:102: Tcl command execution failed: conflict GCCcore
1c. Prepare your reference file
below content from F.Scucchia page
The GATK uses two files to access and safety check access to the reference files: a .dict dictionary of the contig names and sizes, and a .fai fasta index file to allow efficient random access to the reference bases. You have to generate these files in order to be able to use a fasta file as reference.
Step 1: CreateSequenceDictionary.jar from Picard to create a .dict file from a fasta file. This produces a SAM-style header file describing the contents of the fasta file.
Step 2: faidx command in samtools to prepare the fasta index file. This file describes byte offsets in the fasta file for each contig, allowing to compute exactly where a particular reference base at contig:pos is in the fasta file. This produces a text file with one record per line for each of the fasta contigs. Each record is of the: contig, size, location, basesPerLine, bytesPerLine.
I have already have Step 2 done from a previous project: /data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta.fai. The command to run to create a file like this is: samtools faidx $R with $R as the path to reference file.
prep_ref.sh:
Takes under 5 minutes.
#!/bin/sh
#SBATCH -t 200:00:00
#SBATCH --export=NONE
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/SNP
#SBATCH --error=output_messages/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=output_messages/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
# Load modules needed
module load GATK/4.3.0.0-GCCcore-11.2.0-Java-11
R="/data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta"
# Step 1 described in notebook post
gatk CreateSequenceDictionary -REFERENCE $R -OUTPUT ${R%*.*}.dict
Output: Montipora_capitata_HIv3.assembly.dict.
1d. Merge unalinged bam file
Merge the unmapped.rg.bam files
At this point they are all named R1 but contain data from both R1 and R2; see output from 1a.
Purpose: Merge unalinged bam file (now with read group info) with aligned bam file (read group info from unalinged bam is transfered to aligned bam)
Input: ${i}.FastqToSam.unmapped.bam.rg.bam from the previous script (edited merged bam file)
Output: ${i}.MergeBamAlignment.merged.bam ; edited merged bam file
I had trouble figuring out how to do this in an array that would be able to recognize that sample’s aligned bam file. The below is the loop I was trying:
# Create a list for every file that ends with FastqToSam.unmapped.bam.rg.bam in that folder
array1=($(ls $path/*.unmapped.bam.rg.bam))
# Create for loop
for i in ${array1[@]}; do
gatk MergeBamAlignment \
--REFERENCE_SEQUENCE $G \
--UNMAPPED_BAM ${i} \
--ALIGNED_BAM $A/*.bam \
--OUTPUT ${i}.merged.bam \
--INCLUDE_SECONDARY_ALIGNMENTS false \
--VALIDATION_STRINGENCY SILENT touch ${i}.Merge.done
done
MergeBamAlignment.sh:
Run time = ~22 min per sample (x40 = ~14 hours)
#!/bin/sh
#SBATCH -t 200:00:00
#SBATCH --export=NONE
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/merged_bam
#SBATCH --error=../output_messages/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../output_messages/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
module load GATK/4.3.0.0-GCCcore-11.2.0-Java-11
G="/data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta"
A="/data/putnamlab/estrand/BleachingPairs_RNASeq/output2"
# Data path to unmapped bam files reads
path="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/unmapped_bam"
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.16_S121_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.16_S121_L003_R1_001.fastq.gz.bam --OUTPUT 16_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 16_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.17_S122_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.17_S122_L003_R1_001.fastq.gz.bam --OUTPUT 17_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 17_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.21_S123_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.21_S123_L003_R1_001.fastq.gz.bam --OUTPUT 21_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 21_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.22_S124_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.22_S124_L003_R1_001.fastq.gz.bam --OUTPUT 22_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 22_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.23_S125_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.23_S125_L003_R1_001.fastq.gz.bam --OUTPUT 23_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 23_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.24_S126_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.24_S126_L003_R1_001.fastq.gz.bam --OUTPUT 24_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 24_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.25_S127_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.25_S127_L003_R1_001.fastq.gz.bam --OUTPUT 25_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 25_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.26_S128_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.26_S128_L003_R1_001.fastq.gz.bam --OUTPUT 26_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 26_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.28_S129_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.28_S129_L003_R1_001.fastq.gz.bam --OUTPUT 28_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 28_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.29_S130_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.29_S130_L003_R1_001.fastq.gz.bam --OUTPUT 29_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 29_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.2_S118_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.2_S118_L003_R1_001.fastq.gz.bam --OUTPUT 2_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 2_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.30_S131_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.30_S131_L003_R1_001.fastq.gz.bam --OUTPUT 30_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 30_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.31_S132_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.31_S132_L003_R1_001.fastq.gz.bam --OUTPUT 31_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 31_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.33_S133_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.33_S133_L003_R1_001.fastq.gz.bam --OUTPUT 33_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 33_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.37_S134_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.37_S134_L003_R1_001.fastq.gz.bam --OUTPUT 37_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 37_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.39_S135_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.39_S135_L003_R1_001.fastq.gz.bam --OUTPUT 39_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 39_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.42_S136_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.42_S136_L003_R1_001.fastq.gz.bam --OUTPUT 42_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 42_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.43_S137_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.43_S137_L003_R1_001.fastq.gz.bam --OUTPUT 43_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 43_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.45_S138_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.45_S138_L003_R1_001.fastq.gz.bam --OUTPUT 45_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 45_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.46_S139_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.46_S139_L003_R1_001.fastq.gz.bam --OUTPUT 46_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 46_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.47_S140_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.47_S140_L003_R1_001.fastq.gz.bam --OUTPUT 47_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 47_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.4_S119_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.4_S119_L003_R1_001.fastq.gz.bam --OUTPUT 4_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 4_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.50_S141_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.50_S141_L003_R1_001.fastq.gz.bam --OUTPUT 50_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 50_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.51_S142_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.51_S142_L003_R1_001.fastq.gz.bam --OUTPUT 51_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 51_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.52_S143_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.52_S143_L003_R1_001.fastq.gz.bam --OUTPUT 52_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 52_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.54_S144_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.54_S144_L003_R1_001.fastq.gz.bam --OUTPUT 54_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 54_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.55_S145_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.55_S145_L003_R1_001.fastq.gz.bam --OUTPUT 55_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 55_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.56_S146_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.56_S146_L003_R1_001.fastq.gz.bam --OUTPUT 56_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 56_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.57_S147_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.57_S147_L003_R1_001.fastq.gz.bam --OUTPUT 57_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 57_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.59_S148_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.59_S148_L003_R1_001.fastq.gz.bam --OUTPUT 59_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 59_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.60_S149_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.60_S149_L003_R1_001.fastq.gz.bam --OUTPUT 60_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 60_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.61_S150_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.61_S150_L003_R1_001.fastq.gz.bam --OUTPUT 61_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 61_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.62_S151_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.62_S151_L003_R1_001.fastq.gz.bam --OUTPUT 62_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 62_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.63_S152_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.63_S152_L003_R1_001.fastq.gz.bam --OUTPUT 63_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 63_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.64_S153_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.64_S153_L003_R1_001.fastq.gz.bam --OUTPUT 64_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 64_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.65_S154_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.65_S154_L003_R1_001.fastq.gz.bam --OUTPUT 65_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 65_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.66_S155_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.66_S155_L003_R1_001.fastq.gz.bam --OUTPUT 66_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 66_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.67_S156_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.67_S156_L003_R1_001.fastq.gz.bam --OUTPUT 67_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 67_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.68_S157_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.68_S157_L003_R1_001.fastq.gz.bam --OUTPUT 68_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 68_trimmed.MergeBamAlignment.done
gatk MergeBamAlignment --REFERENCE_SEQUENCE $G --UNMAPPED_BAM $path/trimmed.6_S120_L003_R1_001.fastq.gz.FastqToSam.unmapped.bam.rg.bam --ALIGNED_BAM $A/trimmed.6_S120_L003_R1_001.fastq.gz.bam --OUTPUT 6_trimmed.MergeBamAlignment.merged.bam --INCLUDE_SECONDARY_ALIGNMENTS false --VALIDATION_STRINGENCY SILENT; touch 6_trimmed.MergeBamAlignment.done
02. MarkDuplicates
Potential PCR duplicates need to be marked with Picard Tools.
Purpose: Merge read groups belonging to the same sample into a single BAM file.
Input: *.MergeBamAlignment.merged.bam from previous step (within merged_bam folder).
Output: *.MarkDuplicates.dedupped.bam and *.MarkDuplicates.metrics files.
MarkDuplicates.sh:
Run time = ~10 min per sample.
#!/bin/sh
#SBATCH -t 200:00:00
#SBATCH --export=NONE
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/merged_bam
#SBATCH --error=../output_messages/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../output_messages/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
module load GATK/4.3.0.0-GCCcore-11.2.0-Java-11
F="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/merged_bam"
array1=($(ls $F/*MergeBamAlignment.merged.bam))
for i in ${array1[@]}; do
gatk MarkDuplicates --INPUT ${i} \
--OUTPUT ${i}.MarkDuplicates.dedupped.bam \
--CREATE_INDEX true \
--VALIDATION_STRINGENCY SILENT \
--METRICS_FILE ${i}.MarkDuplicates.metrics
done
03. SplitNCigarReads
Content below from F.Succhia protocol.
The ‘CIGAR’ (Compact Idiosyncratic Gapped Alignment Report) string is how the SAM/BAM format represents spliced alignments. Understanding the CIGAR string will help you understand how your query sequence aligns to the reference genome.
Purpose: This will split reads that contain Ns in their cigar string (e.g. spanning splicing events in RNAseq data), it identifies all N cigar elements and creates k+1 new reads (where k is the number of N cigar elements). This is to distinguish between deletions in exons and large skips due to introns. For mRNA-to-genome alignment, an N operation represents an intron.
Input: *.MarkDuplicates.dedupped.bam from previous step.
Output: *.SplitNCigarReads.split.bam file.
SplitNCigarReads.sh:
Run time = ~99 min per sample (x40 = 66 hours; almost 3 days)
#!/bin/sh
#SBATCH -t 300:00:00
#SBATCH --export=NONE
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/SplitNCigarReads
#SBATCH --error=../output_messages/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../output_messages/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
module load GATK/4.3.0.0-GCCcore-11.2.0-Java-11
# List paths for input files (F) and reference genome (G)
F="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/merged_bam"
G="/data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta"
# Create array1 list of all files we want as input
array1=($(ls $F/*MarkDuplicates.dedupped.bam))
# Create for loop for SplitNCigarReads function
for i in ${array1[@]}; do
gatk SplitNCigarReads \
-R $G \
-I ${i} \
-O ${i}.SplitNCigarReads.split.bam
done
Content below from F.Succhia protocol.
Steps 4 and 5 are part of the GATK “Best Practices” guide but can’t really be undertaken with non-model genomes. These steps in fact require “known sites”, i.e. sites where we know beforehand that SNPs occure. This info is not avaliable for non-model systems. Since sites not in this list are considered putative errors that need to be corrected, these steps have to be skipped.
06. HaplotypeCaller
This assumes: –sample-ploidy 2 (default) –heterozygosity 0.001 (deafult; add details.)
This assumes plodiy = 2. Based on our work with M. capitata in Kaneohe Bay, Hawai’i, these populations are diploidy.
Input: *.SplitNCigarReads.split.bam from previous step.
Output: ${i}.HaplotypeCaller.g.vcf.gz file.
HaplotypeCaller.sh:
Run time = 7 days 6 hours
This look longer than expected.. I probably could have upped the # of threads and processors? Once it started I just let it run b/c that week was busy for me anyway.
#!/bin/sh
#SBATCH -t 300:00:00
#SBATCH --export=NONE
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/merged_bam
#SBATCH --error=../output_messages/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../output_messages/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
module load GATK/4.3.0.0-GCCcore-11.2.0-Java-11
# List paths for input files (F) and reference genome (G)
F="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/merged_bam"
G="/data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta"
# Create array1 list of all files we want as input
array1=($(ls $F/*SplitNCigarReads.split.bam))
# Create for loop for HaplotypeCaller function
for i in ${array1[@]}; do
gatk HaplotypeCaller \
--reference $G \
--input ${i} \
--output ${i}.HaplotypeCaller.g.vcf.gz -dont-use-soft-clipped-bases -ERC GVCF
done
move the .vcf output files to a new directory to keeep the outputs clean
mv merged_bam/*vcf.gz vcf into new vcf directory.
mv merged_bam/*tbi vcf into new vcf directory.
So each sample has a vcf.gz file and index vcf.gz.tbi file.
Usually I would also edit the names now to remove the .bam.bam.bam notation but the vcf files in the code above took so long I’ll leave it just in case.
07. Combine *.g.vcf.gz files and call genotypes
If you are generating large callsets (1000+ samples) then look into GenomicsDBImport instead.
Input: *HaplotypeCaller.g.vcf.gz. Two or more HaplotypeCaller GVCFs to combine (only those produced by HaplotypeCaller).
Output: single cohort.g.vcf.gz file. A combined multi-sample gVCF.
https://gatk.broadinstitute.org/hc/en-us/articles/360037053272-CombineGVCFs
I also tried to run these in an array / multiple files after the -V flag, but ran into errors.
Combine GVCF files
CombineGVCFs.sh:
Run time = 2.5 - 3 hrs for 40 samples.
#!/bin/sh
#SBATCH -t 60:00:00
#SBATCH --export=NONE
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/vcf
#SBATCH --error=../output_messages/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../output_messages/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
module load GATK/4.3.0.0-GCCcore-11.2.0-Java-11
F="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/vcf"
G="/data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta"
gatk CombineGVCFs -R "${G}" \
-V 16_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 17_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 21_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 22_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 23_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 24_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 25_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 26_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 28_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 29_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 2_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 30_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 31_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 33_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 37_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 39_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 42_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 43_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 45_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 46_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 47_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 4_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 50_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 51_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 52_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 54_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 55_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 56_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 57_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 59_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 60_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 61_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 62_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 63_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 64_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 65_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 66_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 67_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 68_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-V 6_trimmed.MergeBamAlignment.merged.bam.MarkDuplicates.dedupped.bam.SplitNCigarReads.split.bam.HaplotypeCaller.g.vcf.gz \
-O cohort.g.vcf.gz
Genotype combined GVCF file
https://gatk.broadinstitute.org/hc/en-us/articles/360037057852-GenotypeGVCFs
Input: single cohort.g.vcf.gz file. A combined multi-sample gVCF made above. The GATK4 GenotypeGVCFs tool can take only one input track. Options are 1) a single single-sample GVCF 2) a single multi-sample GVCF created by CombineGVCFs or 3) a GenomicsDB workspace created by GenomicsDBImport. A sample-level GVCF is produced by HaplotypeCaller with the -ERC GVCF setting. Only GVCF files produced by HaplotypeCaller (or CombineGVCFs) can be used as input for this tool.
Output: single cohort_genotypes.vcf.gz file. A final VCF in which all samples have been jointly genotyped.
This portion can handle any type of ploidy (not needed here but for Hawai’i P. acuta it would be).
GenotypeGVCFs.sh:
Run time = 2+ hours - I wasn’t able to catch the exact time of this.
#!/bin/sh
#SBATCH -t 60:00:00
#SBATCH --export=NONE
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/vcf
#SBATCH --error=../output_messages/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../output_messages/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
module load GATK/4.3.0.0-GCCcore-11.2.0-Java-11
F="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/vcf"
G="/data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta"
gatk --java-options "-Xmx4g" GenotypeGVCFs \
--reference "${G}" \
--output "cohort_genotypes.vcf.gz" \
--variant "cohort.g.vcf.gz" \
-stand-call-conf 30
Flags:
--variant=-V(same flag)-stand-call-conf= standard-min-confidence-threshold-for-calling. The minimum phred-scaled confidence threshold at which variants should be called. Default is 10.0. Similar pipelines with coral and our collaborators have used 30 so this is what I’m using for now.--annotationcan be used to pair annotation files. I need to circle back to this to see which file is most appropriate.. Federica had--annotation AS_MappingQualityRankSumTest --annotation AS_ReadPosRankSumTest > GenotypeGVCFs.log 2>&1.
08. Select SNPs and Indels
Variant_Filtration_1.sh:
Argument 1 and 2 from https://github.com/fscucchia/Pastreoides_development_depth/blob/main/SNPs/Variant_Filtration.sh.
Run time = ~ 10 minutes
#!/bin/sh
#SBATCH -t 60:00:00
#SBATCH --export=NONE
#SBATCH --ntasks-per-node=24
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/vcf
#SBATCH --error=../output_messages/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../output_messages/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
module load GATK/4.3.0.0-GCCcore-11.2.0-Java-11
F="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/vcf"
G="/data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta"
OUT="GVCFall"
O="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration"
## Argument 1
gatk SelectVariants \
--reference "${G}" \
--variant "$F/cohort_genotypes.vcf.gz" \
--output "$O/${OUT}_SNPs.vcf.gz" \
-select-type SNP 1> "$O/${OUT}_SNPs.vcf.gz.log" 2>&1
gatk SelectVariants \
--reference "${G}" \
--variant "cohort_genotypes.vcf.gz" \
--output "$O/${OUT}_INDELs.vcf.gz" \
-select-type INDEL 1> "$O/${OUT}_INDELs.vcf.gz.log" 2>&1
## Argument 2
gatk VariantsToTable \
--reference "${G}" \
--variant "$O/${OUT}_SNPs.vcf.gz" \
--output "$O/${OUT}_SNPs.table" \
-F CHROM -F POS -F QUAL -F QD -F DP -F MQ -F FS -F SOR -F MQRankSum -F ReadPosRankSum 1> "$O/${OUT}_SNPs.table.log" 2>&1
gatk VariantsToTable \
--reference "${G}" \
--variant "$O/${OUT}_INDELs.vcf.gz" \
--output "$O/${OUT}_INDELs.table" \
-F CHROM -F POS -F QUAL -F QD -F DP -F MQ -F FS -F SOR -F MQRankSum -F ReadPosRankSum 1> "$O/${OUT}_INDELs.table.log" 2>&1
Switch to R
Copy GVCFall_SNPs.table and GVCFall_INDELs.table to repo.
scp emma_strand@ssh3.hac.uri.edu:/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration/GVCFall_SNPs.table /Users/emmastrand/MyProjects/HI_Bleaching_Timeseries/Dec-July-2019-analysis/output/WGBS/SNP
scp emma_strand@ssh3.hac.uri.edu:/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration/GVCFall_INDELs.table /Users/emmastrand/MyProjects/HI_Bleaching_Timeseries/Dec-July-2019-analysis/output/WGBS/SNP
Run VariantFiltering.Rmd Part 1 script to create diagnostic plots.
- https://support.illumina.com/content/dam/illumina-support/help/Illumina_DRAGEN_Bio_IT_Platform_v3_7_1000000141465/Content/SW/Informatics/Dragen/QUAL_QD_GQ_Formulation_fDG.htm
- Slide 19 has GATK recommendations for non model organisms: https://ressources.france-bioinformatique.fr/sites/default/files/V04_FiltrageVariantNLapaluRoscoff2016_0.pdf
-
https://gatk.broadinstitute.org/hc/en-us/articles/360035890471-Hard-filtering-germline-short-variants#:~:text=StrandOddsRatio%20(SOR),-This%20is%20another&text=Reads%20at%20the%20ends%20of,0%20to%20greater%20than%209.
QD= quality (QUAL) normalized by the read depth (DP) or Variant Quality / depth of non-ref samplesDP= read depth / Coverage (reads that passed quality metrics)FS= Test (Fisher) : Phred score p-value for strand biasMQ= Root Mean Square Mapping QualityMQRankSum= Mapping quality of Reference reads vs ALT readsReadPosRankSum= Distance of ALT reads from the end of the readsSOR= StrandOddsRatio; Reads at the ends of exons tend to only be covered by reads in one direction and FS gives those variants a bad score. SOR will take into account the ratios of reads that cover both alleles. Let’s look at the SOR values for the unfiltered variants. The SOR values range from 0 to greater than 9.

09. Apply Variant filtering
https://gatk.broadinstitute.org/hc/en-us/articles/360050815032-VariantFiltration
https://github.com/fscucchia/Pastreoides_development_depth/blob/main/SNPs/Variant_filt.sh
Parameters chosen from the diagnostic plots above. I compared the GATK recommended minimum filtering to my plots and to what Federica did for her Porites spp. filtering. These filters are fairly conservative so I could come back and loosen them if needed.
Variant_Filtration_2.sh:
Run time <5 min
#!/bin/sh
#SBATCH -t 60:00:00
#SBATCH --export=NONE
#SBATCH --ntasks-per-node=24
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration
#SBATCH --error=../output_messages/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../output_messages/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
module load GATK/4.3.0.0-GCCcore-11.2.0-Java-11
G="/data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta"
OUT="GVCFall"
O="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration"
###
SNP_QD_MIN=20.000
SNP_MQ_MIN=50.000
SNP_FS_MAX=5.000
SNP_SOR_MAX=2.500
####
gatk VariantFiltration --reference "${G}" --variant "${OUT}_SNPs.vcf.gz" --output "$O/${OUT}_SNPs_VarScores_filter.vcf.gz" \
--filter-name "VarScores_filter_QD" --filter-expression "QD < $SNP_QD_MIN" \
--filter-name "VarScores_filter_MQ" --filter-expression "MQ < $SNP_MQ_MIN" \
--filter-name "VarScores_filter_FS" --filter-expression "FS > $SNP_FS_MAX" \
--filter-name "VarScores_filter_SOR" --filter-expression "SOR > $SNP_SOR_MAX" \
1> "$O/${OUT}_SNPs_VarScores_filter.vcf.gz.log" 2>&1
Check number PASSED after the first filtering
Move to Variant_Filtration directory.
$ zcat "GVCFall_SNPs.vcf.gz" | grep -v '^#' | wc -l
## output:
3970239
$ zcat "GVCFall_SNPs_VarScores_filter.vcf.gz" | grep 'PASS' | wc -l
## output:
2485639
After filtering, 62% of SNPs passed (2485639/3970239). Variant_Filtration_2.sh doesn’t remove these yet, only annotates with pass or fail.
Extract only variants that PASSED filtering
Move to Variant_Filtration directory.
zcat "GVCFall_SNPs_VarScores_filter.vcf.gz" | grep -E '^#|PASS' > "GVCFall_SNPs_VarScores_filterPASSED.vcf"
interactive
module load GATK/4.3.0.0-GCCcore-11.2.0-Java-11
gatk IndexFeatureFile --input "GVCFall_SNPs_VarScores_filterPASSED.vcf" 1> "GVCFall_SNPs_VarScores_filterPASSED.vcf.log" 2>&1
Check filtering worked
Variant_Filtration_3.sh:
Run time <5 min
#!/bin/sh
#SBATCH -t 60:00:00
#SBATCH --export=NONE
#SBATCH --ntasks-per-node=24
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration
#SBATCH --error=../output_messages/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../output_messages/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
module load GATK/4.3.0.0-GCCcore-11.2.0-Java-11
G="/data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta"
OUT="GVCFall"
O="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration"
gatk VariantsToTable --reference "${G}" --variant "${OUT}_SNPs_VarScores_filterPASSED.vcf" \
--output "${OUT}_SNPs_VarScores_filterPASSED.table" \
-F CHROM -F POS -F QUAL -F QD -F DP -F MQ -F FS -F SOR -F MQRankSum -F ReadPosRankSum \
1> "${OUT}_SNPs_VarScores_filterPASSED.table.log" 2>&1
Copy GVCFall_SNPs_VarScores_filterPASSED.table to repo.
scp emma_strand@ssh3.hac.uri.edu:/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration/GVCFall_SNPs_VarScores_filterPASSED.table /Users/emmastrand/MyProjects/HI_Bleaching_Timeseries/Dec-July-2019-analysis/output/WGBS/SNP
Run VariantFiltering.Rmd Part 2 script to create diagnostic plots.

This worked!
2nd-pass filtering, filter genotypes part 1
When all low confidence variant sites are removed, filter VCF files for genotype quality.
Variant_Filtration_4.sh:
Run time < 10 minutes
#!/bin/sh
#SBATCH -t 60:00:00
#SBATCH --export=NONE
#SBATCH --ntasks-per-node=24
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration
#SBATCH --error=../output_messages/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../output_messages/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
module load GATK/4.3.0.0-GCCcore-11.2.0-Java-11
G="/data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta"
OUT="GVCFall"
O="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration"
F="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/vcf"
gatk VariantsToTable \
--reference "${G}" \
--variant "$F/cohort_genotypes.vcf.gz" \
--output "${OUT}.DP.table" \
-F CHROM -F POS -GF GT -GF DP 1> "${OUT}.DP.table.log" 2>&1
Flags:
GF/--genotype-fields= The name of a genotype field to include in the output table (“DP” and “GT”)F/--fields= The name of a standard VCF field or an INFO field to include in the output table
GT = genotype DP = read depth / Coverage (reads that passed quality metrics)
From all samples before previous filtering. Run the following command:
for ((i=3; i<=81; i +=2)); do cut -f $i,$((i+1)) GVCFall.DP.table | awk '$1 != "./." {print $2}' > $i.DP; done
Where 3-81 are column numbers. We only want the odd numbers for 40 samples (column 3, 5, 7, etc) and skipping the even. This numbering is required because every sample is represented by two columns (GT, DP). Split it by samples and keep only positions that have been genotyped (!=”./.”). Output is a file per sample ($i.DP) with the column # as the ID ($i).
Copy output of the above command $i.DP to repo. Make new directory ‘DP’ to make scp -r easier.
scp -r emma_strand@ssh3.hac.uri.edu:/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration/DP /Users/emmastrand/MyProjects/HI_Bleaching_Timeseries/Dec-July-2019-analysis/output/WGBS/SNP
Run VariantFiltering.Rmd Part 3 script to create DP distribution plots.

View the content of GVCFall.DP.percentiles.txt and select the acceptable cut-off. Discard the genotypes below the 5th percentile and above the 99th percentile.
Come back to how to do the above 5% and 99% …
2nd-pass filtering, filter genotypes part 2: apply dp filtering and set no calls
Input: ${OUT}_SNPs_VarScores_filterPASSED.vcf
Ouput: ${OUT}_SNPs_VarScores_filterPASSED_DPfilter.vcf and ${OUT}_SNPs_VarScores_filterPASSED_DPfilterNoCall.vcf
Variant_Filtration_5.sh:
Run time < 10 minutes
#!/bin/sh
#SBATCH -t 60:00:00
#SBATCH --export=NONE
#SBATCH --ntasks-per-node=24
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration
#SBATCH --error=../output_messages/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../output_messages/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
module load GATK/4.3.0.0-GCCcore-11.2.0-Java-11
G="/data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta"
OUT="GVCFall"
O="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration"
F="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/vcf"
###
DP_MIN=20.000
###
gatk VariantFiltration \
--reference "${G}" \
--variant "${OUT}_SNPs_VarScores_filterPASSED.vcf" \
--output "${OUT}_SNPs_VarScores_filterPASSED_DPfilter.vcf" \
--genotype-filter-name "DP_filter" \
--genotype-filter-expression "DP < $DP_MIN" \
1> "${OUT}_SNPs_VarScores_filterPASSED_DPfilter.vcf.log" 2>&1
gatk SelectVariants \
--reference "${G}" \
--variant "${OUT}_SNPs_VarScores_filterPASSED_DPfilter.vcf" \
--output "${OUT}_SNPs_VarScores_filterPASSED_DPfilterNoCall.vcf" \
--set-filtered-gt-to-nocall \
1> "${OUT}_SNPs_VarScores_filterPASSED_DPfilterNoCall.vcf.log" 2>&1
3nd-pass filtering
Input: ${OUT}_SNPs_VarScores_filterPASSED_DPfilterNoCall.vcf
Output: ${OUT}_SNPs_VarScores_filterPASSED_DPfilterNoCall.AD.table
Variant_Filtration_6.sh:
Run time < 10 minutes
#!/bin/sh
#SBATCH -t 60:00:00
#SBATCH --export=NONE
#SBATCH --ntasks-per-node=24
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration
#SBATCH --error=../output_messages/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../output_messages/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
module load GATK/4.3.0.0-GCCcore-11.2.0-Java-11
G="/data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta"
OUT="GVCFall"
O="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration"
F="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/vcf"
gatk VariantsToTable \
--reference "${G}" \
--variant "${OUT}_SNPs_VarScores_filterPASSED_DPfilterNoCall.vcf" \
--output "${OUT}_SNPs_VarScores_filterPASSED_DPfilterNoCall.AD.table" \
-F CHROM -F POS -GF GT -GF AD -GF DP \
1> "${OUT}_SNPs_VarScores_filterPASSED_DPfilterNoCall.AD.table.log" 2>&1
10. VCF to Table
Variant_Filtration_7.sh:
#!/bin/sh
#SBATCH -t 60:00:00
#SBATCH --export=NONE
#SBATCH --ntasks-per-node=24
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration
#SBATCH --error=../output_messages/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../output_messages/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
module load GATK/4.3.0.0-GCCcore-11.2.0-Java-11
G="/data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta"
OUT="GVCFall"
O="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration"
F="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/vcf"
gatk VariantsToTable \
--reference "${G}" \
--variant "${OUT}_SNPs_VarScores_filterPASSED_DPfilterNoCall.vcf" \
--output "${OUT}_SNPs_filterPASSED_final.table" \
-F CHROM -F POS -GF GT \
1> "${OUT}_SNPs_filterPASSED_final.table.log" 2>&1
Output: ${OUT}_SNPs_filterPASSED_final.table. Output files (genotype tables) and vcf files ready for subsequent analyses.
11. Filtering for linkage disequilibrium
Plink2 program: https://www.cog-genomics.org/plink/2.0/general_usage. Program commands start with plink2.
Argument 1: Which manually sets the variant ID in the final vcf file (it replaces the dot in the ID column with a manually set ID). This is needed for argument 4 to work.
Argument 2: Which converts the vcf file into a bed file.
--double-id: deals with Multiple instances of ‘_’ in sample ID--max-alleles 2: allows for multiallelic variants--chr-set: sets # of chromosomes (28) withno-xyoption
Argument 3: Which performs the pruning of SNPs that are strongly genetically linked.
--allow-no-sexno longer has any effect in the newest version so don’t need this.--bad-ldwasn’t recognized so left this out too.
Argument 4: Which extracts the pruned SNPs data from the vcf file.
--max-alleles 2: allows for multiallelic variants
plink.sh:
#!/bin/sh
#SBATCH -t 60:00:00
#SBATCH --export=NONE
#SBATCH --ntasks-per-node=24
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration
#SBATCH --error=../output_messages/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../output_messages/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
module load BCFtools/1.9-foss-2018b
module load PLINK/2.00a2.3_x86_64
G="/data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta"
OUT="GVCFall"
F="/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration"
## Argument 1
bcftools annotate --set-id '%CHROM\_%POS\_%REF\_%FIRST_ALT' \
$F/${OUT}_SNPs_VarScores_filterPASSED_DPfilterNoCall.vcf
## Argument 2
plink2 --vcf $F/${OUT}_SNPs_VarScores_filterPASSED_DPfilterNoCall.vcf \
--make-bed --chr-set 28 no-xy --allow-extra-chr --double-id --max-alleles 2 --out VCF_annotated
## Argument 3
plink2 --bfile $F/VCF_annotated \
--allow-extra-chr --indep-pairwise 25 0.5
## Argument 4
plink2 --vcf $F/${OUT}_SNPs_VarScores_filterPASSED_DPfilterNoCall.vcf \
--extract plink2.prune.in --allow-extra-chr --make-bed --double-id --max-alleles 2 --out final_vcf_pruned
Output message:
=40 samples (0 females, 0 males, 40 ambiguous; 40 founders) loaded from
final_vcf_pruned-temporary.psam.
2471727 out of 2485639 variants loaded from final_vcf_pruned-temporary.pvar.
Note: No phenotype data present.
--extract: 2471727 variants remaining.
2471727 variants remaining after main filters.
12. Fst analysis
Fst.Rmd R script ran from the below files copied from the output.
scp emma_strand@ssh3.hac.uri.edu:/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration/final_vcf_pruned.bed /Users/emmastrand/MyProjects/HI_Bleaching_Timeseries/Dec-July-2019-analysis/output/WGBS/SNP/
scp emma_strand@ssh3.hac.uri.edu:/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration/final_vcf_pruned.fam /Users/emmastrand/MyProjects/HI_Bleaching_Timeseries/Dec-July-2019-analysis/output/WGBS/SNP/
scp emma_strand@ssh3.hac.uri.edu:/data/putnamlab/estrand/BleachingPairs_RNASeq/SNP/Variant_Filtration/final_vcf_pruned.bim /Users/emmastrand/MyProjects/HI_Bleaching_Timeseries/Dec-July-2019-analysis/output/WGBS/SNP/