
KBay Bleaching Pairs WGBS Analysis Pipeline
KBAY WGBS Methylation Analysis Pipeline
Raw data folder path: /data/putnamlab/KITT/hputnam/20211008_BleachingPairs_WGBS
Processed data folder path: /data/putnamlab/estrand/BleachingPairs_WGBS
References:
- Wong and Strand WGBS pipeline info: https://github.com/Putnam-Lab/Lab_Management/blob/master/Bioinformatics_%26_Coding/Workflows/Methylation_QC.md
- Wong Past pipeline: https://github.com/kevinhwong1/Thermal_Transplant_Molecular/blob/main/scripts/Past_WGBS_Workflow.md
- Wong methylseq trimming tests pipeline: https://kevinhwong1.github.io/KevinHWong_Notebook/Methylseq-trimming-test-to-remove-m-bias/
Download genome file from Rutgers
http://cyanophora.rutgers.edu/montipora/ and upload to andromeda.
Contents:
- Setting Up Andromeda
- Initial fastqc on files
- Initial Multiqc Report
- NF-core: Methylseq
- Bismark Multiqc Report
- Merge Strands
- Sort CpG .cov file
- Create bedgraph files
- Filter for a specific coverage (5X, 10X)
- Create a file with positions found in all samples
- Gene Annotation file
- IntersectBed: Loci mapped to annotated gene
- IntersectBed: File to only positions found in all samples
- Exporting Files
Setting Up Andromeda
Make a new directory for output files
$ mkdir BleachingPairs_WGBS
$ mkdir fastqc_results
$ mkdir scripts
Download genome file from Rutgers
http://cyanophora.rutgers.edu/montipora/
Creating a test run folder
Creating a test set folder in case I need to run different iterations of the methylseq script.
$ mkdir test_set
$ cp /data/putnamlab/KITT/hputnam/20211008_BleachingPairs_WGBS/16_S138_L003_R{1,2}_001.fastq.gz /data/putnamlab/estrand/BleachingPairs_WGBS/test_set
$ cp /data/putnamlab/KITT/hputnam/20211008_BleachingPairs_WGBS/17_S134_L003_R{1,2}_001.fastq.gz /data/putnamlab/estrand/BleachingPairs_WGBS/test_set
$ cp /data/putnamlab/KITT/hputnam/20211008_BleachingPairs_WGBS/18_S154_L003_R{1,2}_001.fastq.gz /data/putnamlab/estrand/BleachingPairs_WGBS/test_set
$ cp /data/putnamlab/KITT/hputnam/20211008_BleachingPairs_WGBS/21_S119_L003_R{1,2}_001.fastq.gz /data/putnamlab/estrand/BleachingPairs_WGBS/test_set
$ cp /data/putnamlab/KITT/hputnam/20211008_BleachingPairs_WGBS/22_S120_L003_R{1,2}_001.fastq.gz /data/putnamlab/estrand/BleachingPairs_WGBS/test_set
Initial fastqc on files
fastqc.sh. This took 14 hours for 40 files. Maybe I would do this only on a subset next time.
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=emma_strand@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_WGBS
#SBATCH --error="script_error_fastqc" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_fastqc" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
cd /data/putnamlab/estrand/BleachingPairs_WGBS
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
for file in /data/putnamlab/KITT/hputnam/20211008_BleachingPairs_WGBS/*fastq.gz
do
fastqc $file --outdir /data/putnamlab/estrand/BleachingPairs_WGBS/fastqc_results
done
multiqc --interactive fastqc_results
The first time I ran this I didn’t have the right output path for multiqc because I did ‘cd’ to the wrong folder. This has been fixed in the script above and I ran the multiqc function
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/BleachingPairs_WGBS/multiqc_report.html /Users/emmastrand/MyProjects/HI_Bleaching_Timeseries/Dec-July-2019-analysis/output/WGBS/initial_multiqc_report.html
Initial MultiQC Report
All samples have sequences of a single length (151bp).
| Sample Name | % Dups | % GC | M Seqs |
|---|---|---|---|
| 16_S138_L003_R1_001 | 20.5% | 25% | 74.5 |
| 16_S138_L003_R2_001 | 19.6% | 26% | 74.5 |
| 17_S134_L003_R1_001 | 18.6% | 25% | 65.0 |
| 17_S134_L003_R2_001 | 17.1% | 25% | 65.0 |
| 18_S154_L003_R1_001 | 18.6% | 25% | 72.9 |
| 18_S154_L003_R2_001 | 19.1% | 25% | 72.9 |
| 21_S119_L003_R1_001 | 21.1% | 25% | 74.7 |
| 21_S119_L003_R2_001 | 20.6% | 25% | 74.7 |
| 22_S120_L003_R1_001 | 17.3% | 24% | 55.0 |
| 22_S120_L003_R2_001 | 16.6% | 24% | 55.0 |
| 23_S141_L003_R1_001 | 24.3% | 23% | 69.3 |
| 23_S141_L003_R2_001 | 23.8% | 24% | 69.3 |
| 24_S147_L003_R1_001 | 15.4% | 24% | 48.4 |
| 24_S147_L003_R2_001 | 15.7% | 25% | 48.4 |
| 25_S148_L003_R1_001 | 21.6% | 25% | 137.5 |
| 25_S148_L003_R2_001 | 21.0% | 25% | 137.5 |
| 26_S121_L003_R1_001 | 15.5% | 25% | 48.8 |
| 26_S121_L003_R2_001 | 15.1% | 25% | 48.8 |
| 28_S122_L003_R1_001 | 20.3% | 25% | 93.6 |
| 28_S122_L003_R2_001 | 19.6% | 25% | 93.6 |
| 29_S153_L003_R1_001 | 25.0% | 24% | 105.4 |
| 29_S153_L003_R2_001 | 24.4% | 24% | 105.4 |
| 2_S128_L003_R1_001 | 21.9% | 24% | 77.3 |
| 2_S128_L003_R2_001 | 21.2% | 24% | 77.3 |
| 30_S124_L003_R1_001 | 18.9% | 24% | 68.3 |
| 30_S124_L003_R2_001 | 18.1% | 24% | 68.3 |
| 31_S127_L003_R1_001 | 13.1% | 25% | 21.8 |
| 31_S127_L003_R2_001 | 13.0% | 25% | 21.8 |
| 32_S130_L003_R1_001 | 21.7% | 23% | 72.7 |
| 32_S130_L003_R2_001 | 21.1% | 23% | 72.7 |
| 33_S142_L003_R1_001 | 16.3% | 24% | 53.0 |
| 33_S142_L003_R2_001 | 15.7% | 24% | 53.0 |
| 34_S136_L003_R1_001 | 16.0% | 24% | 60.1 |
| 34_S136_L003_R2_001 | 15.2% | 24% | 60.1 |
| 35_S137_L003_R1_001 | 22.6% | 24% | 84.9 |
| 35_S137_L003_R2_001 | 21.6% | 24% | 84.9 |
| 37_S140_L003_R1_001 | 18.1% | 24% | 64.7 |
| 37_S140_L003_R2_001 | 17.1% | 24% | 64.7 |
| 38_S129_L003_R1_001 | 14.8% | 23% | 49.9 |
| 38_S129_L003_R2_001 | 14.7% | 23% | 49.9 |
| 39_S145_L003_R1_001 | 19.6% | 24% | 75.3 |
| 39_S145_L003_R2_001 | 19.2% | 24% | 75.3 |
| 40_S135_L003_R1_001 | 21.5% | 25% | 82.4 |
| 40_S135_L003_R2_001 | 20.8% | 25% | 82.4 |
| 41_S151_L003_R1_001 | 19.1% | 26% | 70.8 |
| 41_S151_L003_R2_001 | 19.3% | 26% | 70.8 |
| 42_S131_L003_R1_001 | 24.0% | 26% | 88.6 |
| 42_S131_L003_R2_001 | 23.6% | 26% | 88.6 |
| 43_S143_L003_R1_001 | 20.6% | 24% | 90.6 |
| 43_S143_L003_R2_001 | 20.1% | 25% | 90.6 |
| 44_S125_L003_R1_001 | 19.5% | 25% | 68.2 |
| 44_S125_L003_R2_001 | 18.9% | 25% | 68.2 |
| 45_S156_L003_R1_001 | 16.7% | 25% | 68.5 |
| 45_S156_L003_R2_001 | 16.8% | 25% | 68.5 |
| 46_S133_L003_R1_001 | 17.6% | 25% | 62.2 |
| 46_S133_L003_R2_001 | 17.2% | 25% | 62.2 |
| 47_S155_L003_R1_001 | 17.9% | 25% | 75.5 |
| 47_S155_L003_R2_001 | 17.5% | 25% | 75.5 |
| 4_S146_L003_R1_001 | 21.7% | 25% | 104.3 |
| 4_S146_L003_R2_001 | 21.4% | 25% | 104.3 |
| 50_S139_L003_R1_001 | 21.9% | 24% | 89.8 |
| 50_S139_L003_R2_001 | 21.1% | 24% | 89.8 |
| 51_S126_L003_R1_001 | 18.1% | 26% | 61.0 |
| 51_S126_L003_R2_001 | 17.6% | 26% | 61.0 |
| 52_S150_L003_R1_001 | 19.1% | 25% | 62.5 |
| 52_S150_L003_R2_001 | 19.4% | 26% | 62.5 |
| 54_S144_L003_R1_001 | 17.7% | 24% | 82.3 |
| 54_S144_L003_R2_001 | 17.4% | 24% | 82.3 |
| 55_S123_L003_R1_001 | 20.8% | 26% | 100.2 |
| 55_S123_L003_R2_001 | 20.1% | 26% | 100.2 |
| 56_S149_L003_R1_001 | 15.1% | 24% | 50.3 |
| 56_S149_L003_R2_001 | 14.9% | 24% | 50.3 |
| 57_S152_L003_R1_001 | 18.8% | 24% | 84.3 |
| 57_S152_L003_R2_001 | 18.5% | 24% | 84.3 |
| 58_S157_L003_R1_001 | 21.3% | 25% | 60.8 |
| 58_S157_L003_R2_001 | 21.2% | 26% | 60.8 |
| 59_S158_L003_R1_001 | 14.7% | 24% | 61.0 |
| 59_S158_L003_R2_001 | 14.8% | 24% | 61.0 |
| 6_S132_L003_R1_001 | 21.1% | 24% | 80.3 |
| 6_S132_L003_R2_001 | 20.4% | 24% | 80.3 |

Sample 31 appears to have a very low # of reads. Might have to take this out later?



Based on the above, the %reads appear to stabilized ~15 bp.

This peak has a normal distribution (good, what we’re looking for) but is shifted to a peak ~20-25… This is usually higher? Red flag?
Is this shifted because methylation changes unmethylated Cytosine to Thymine and therefore a smaller amount of GC content? Especially in an invertebrate that only has 20% methylation.. This is actually a green flag then?



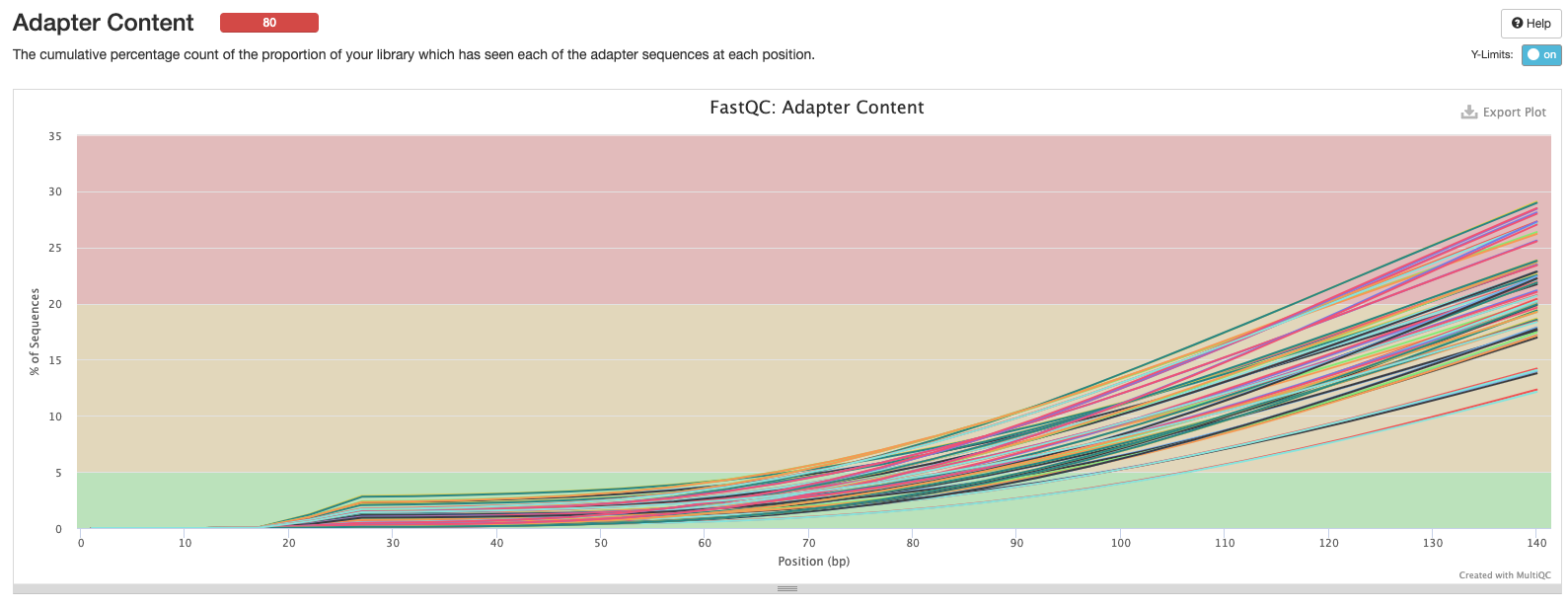
This throws a red flag for me… That seems like a lot of adapter content in places it is not usually?
Does bisulfite conversion cause a higher # of T’s (mostly unmethylated in a WGBS set) that could look like adapters?

NF-core: Methylseq
Goal: Reduce M-bias but keep as much of the sequence as possible. The first iteration of my methylseq script output looked good
Nextflow version 21.03.0 requires an -input command.
The –name output needs to be different every time you run this script.
BleachingPairs_methylseq (1) script: GENOME VERSION 2
nano BleachingPairs_methylseq.sh
#!/bin/bash
#SBATCH --job-name="methylseq"
#SBATCH -t 120:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --mem=100GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_WGBS
#SBATCH -p putnamlab
#SBATCH --cpus-per-task=3
# load modules needed
source /usr/share/Modules/init/sh # load the module function
module load Nextflow/21.03.0
# run nextflow methylseq
nextflow run nf-core/methylseq -profile singularity \
--aligner bismark \
--igenomes_ignore \
--fasta /data/putnamlab/estrand/Montipora_capitata_HIv2.assembly.fasta \
--save_reference \
--input '/data/putnamlab/KITT/hputnam/20211008_BleachingPairs_WGBS/*_R{1,2}_001.fastq.gz' \
--clip_r1 10 \
--clip_r2 10 \
--three_prime_clip_r1 10 --three_prime_clip_r2 10 \
--non_directional \
--cytosine_report \
--relax_mismatches \
--unmapped \
--outdir /data/putnamlab/estrand/BleachingPairs_WGBS \
-name WGBS_methylseq_BleachingPairs3
Ran first and then moved all output to BleachingPairs_methylseq directory folder.
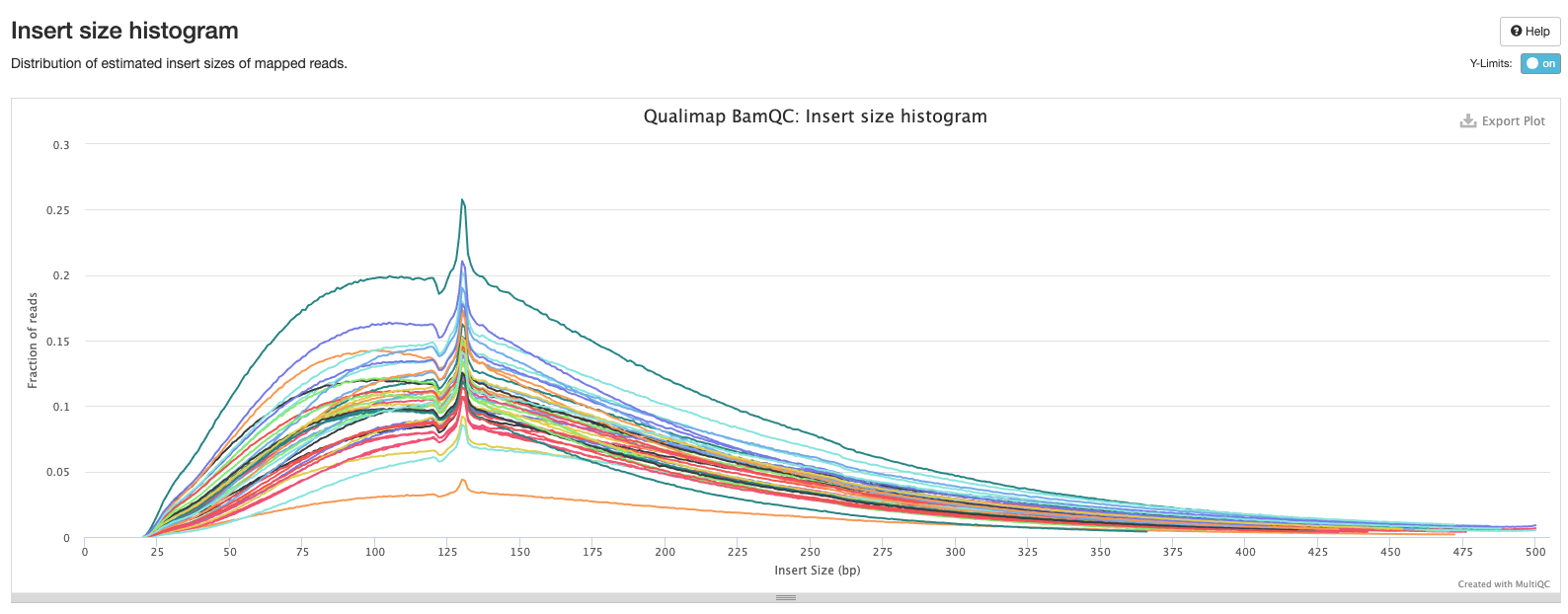
Bismark Multiqc Report
Comparing statistics for the methylseq summary output in this post: https://github.com/hputnam/HI_Bleaching_Timeseries/blob/main/Dec-July-2019-analysis/scripts/methylseq_statistics.md. It does not appear that extraction or pico methyl seq date affected these statistics. I’m good to move on to the following scripts for DMG analysis.
Versions of all packages that bismark uses are in bismark multiqc report output.
Based on the below stats, it looks like the methylseq script (BleachingPairs_methylseq - 1) worked well and there is not an m-bias issue to worry about.
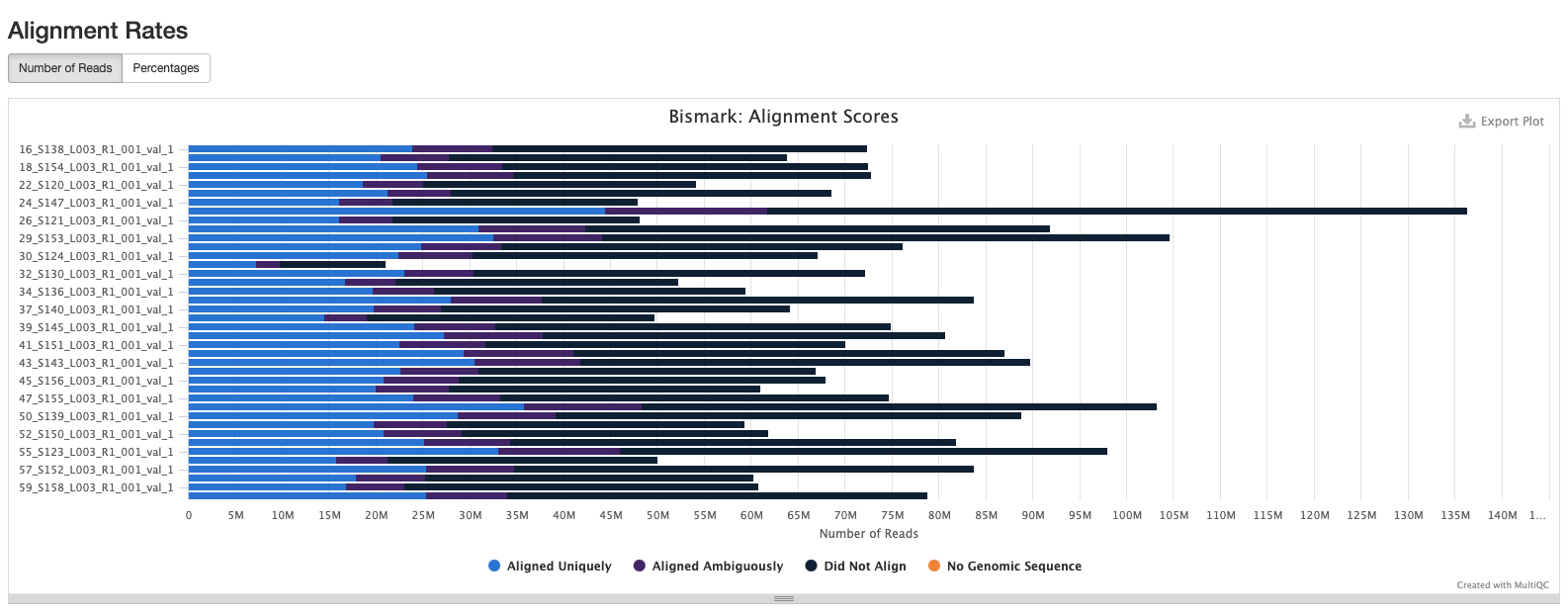

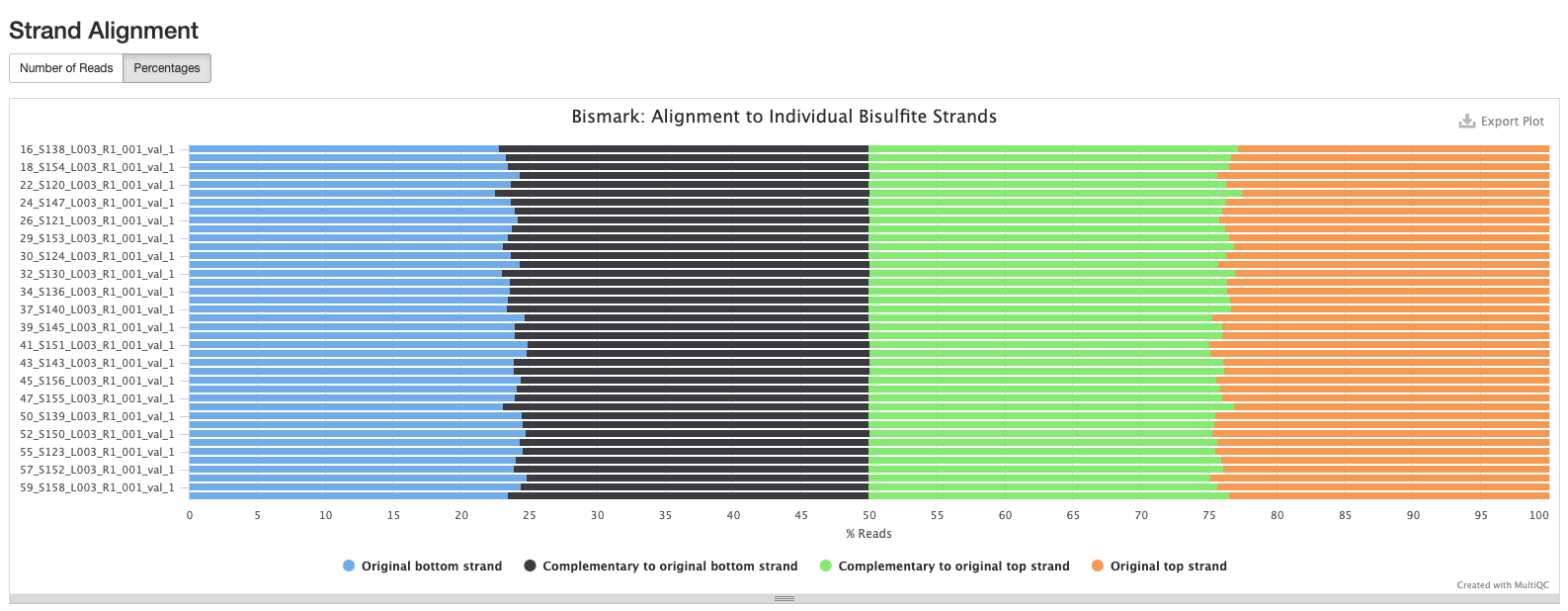

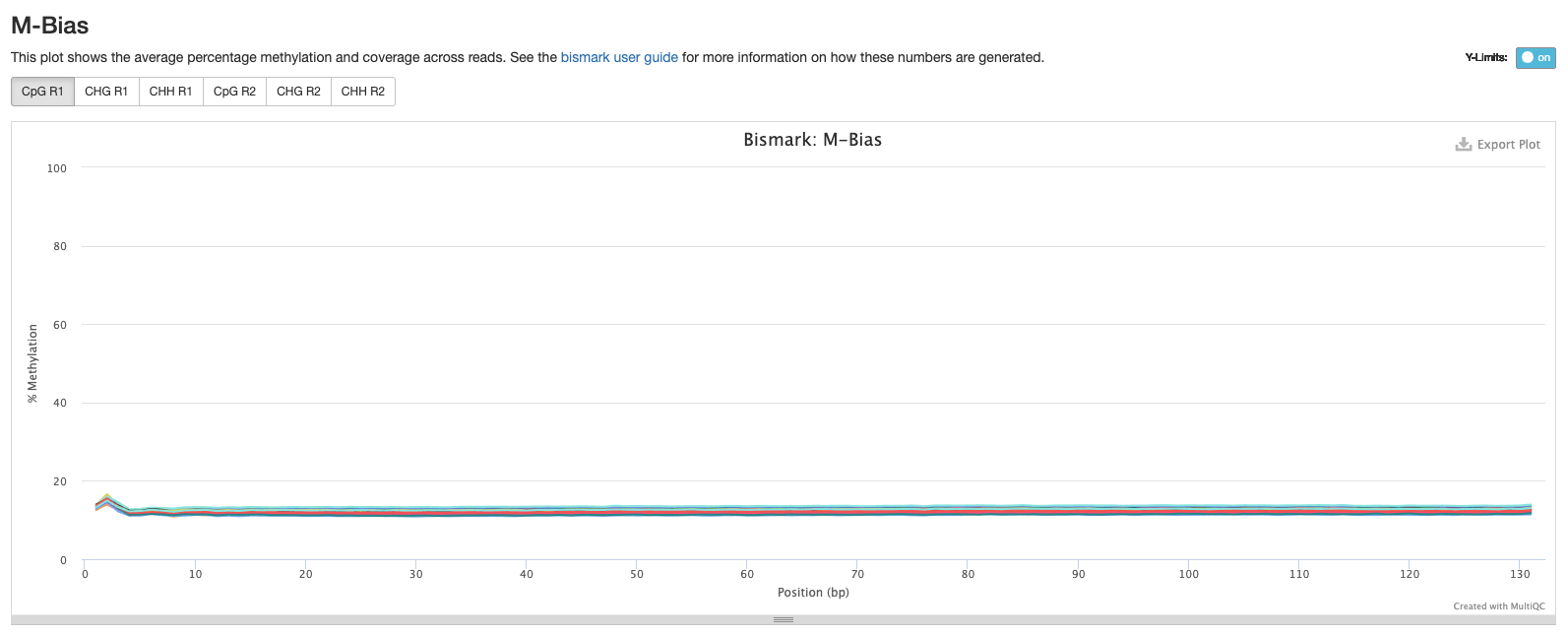






GENOME VERSION 3
wget http://cyanophora.rutgers.edu/montipora/Montipora_capitata_HIv3.assembly.fasta.gz and gunzip Montipora_capitata_HIv3.assembly.fasta.gz.
BleachingPairs_methylseq_v3.sh:
#!/bin/bash
#SBATCH --job-name="v3_BPmeth"
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --mem=100GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_WGBS
#SBATCH --cpus-per-task=3
#SBATCH --error="script_error_v3_BPmeth" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_v3_BPmeth" #once your job is completed, any final job report comments will be put in this file
# load modules needed
source /usr/share/Modules/init/sh # load the module function
module load Nextflow/21.03.0
# run nextflow methylseq
nextflow run nf-core/methylseq -resume \
-profile singularity \
--aligner bismark \
--igenomes_ignore \
--fasta /data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta \
--save_reference \
--input '/data/putnamlab/KITT/hputnam/20211008_BleachingPairs_WGBS/*_R{1,2}_001.fastq.gz' \
--clip_r1 10 \
--clip_r2 10 \
--three_prime_clip_r1 10 --three_prime_clip_r2 10 \
--non_directional \
--cytosine_report \
--relax_mismatches \
--unmapped \
--outdir /data/putnamlab/estrand/BleachingPairs_WGBS/BleachingPairs_methylseq_v3 \
-name WGBS_methylseq_BleachingPairs_v3_3 # this line was taken out later -- not needed
The multiqc function is running into errors from the above script so I ran:
interactive
module load MultiQC/1.9-intel-2020a-Python-3.8.2
multiqc -f --filename WGBS_methylseq_KBAY_genomev3_multiqc_report . \
-m custom_content -m picard -m qualimap -m bismark -m samtools -m preseq -m cutadapt -m fastqc
Copying this file to project folder:
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/BleachingPairs_WGBS/BleachingPairs_methylseq_v3/WGBS_methylseq_KBAY_genomev3_multiqc_report.html /Users/emmastrand/MyProjects/HI_Bleaching_Timeseries/Dec-July-2019-analysis/output/WGBS/WGBS_methylseq_KBAY_genomev3_multiqc_report.html
Originally this was interrupted from storage issues but I re-ran the script with -resume, but output was a second folder:
I ran the script above again to make sure it wasn’t because it was stopped and started several times b/c of storage issues on andromeda. New output will be in /data/putnamlab/estrand/BleachingPairs_WGBS/BleachingPairs_methylseq_v3_2.
Sanity check that all samples ran:
ls bismark_deduplicated/*bam | wc: 40 40 2317
ls bismark_methylation_calls/methylation_coverage/*deduplicated.bismark.cov.gz | wc: 40 40 4637
Multiqc Report
Full report can be found here: https://github.com/hputnam/HI_Bleaching_Timeseries/blob/main/Dec-July-2019-analysis/output/WGBS/WGBS_methylseq_KBAY_genomev3_multiqc_report.html.
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/BleachingPairs_WGBS/BleachingPairs_methylseq_v3_2/MultiQC/multiqc_report.html /Users/emmastrand/MyProjects/HI_Bleaching_Timeseries/Dec-July-2019-analysis/output/WGBS/WGBS_methylseq_KBAY_genomev3_2_multiqc_report.html
Merge Strands
See more detailed information on these steps, what the script is doing, and what the flags mean here:
- https://github.com/emmastrand/EmmaStrand_Notebook/blob/master/_posts/2021-10-21-HoloInt-WGBS-Analysis-Pipeline.md#-merge-strands
- https://github.com/Putnam-Lab/Lab_Management/blob/master/Bioinformatics_%26_Coding/Workflows/Methylation_QC.md#-merge-strands
Input: *deduplicated.bismark.cov.gz.
Output: *merged_CpG_evidence.cov.
This takes 10+ hours (40 samples but # of reads more relevant to this #).
GENOME VERSION 2
Make a new directory for this output: mkdir merged_cov.
File path: /data/putnamlab/estrand/BleachingPairs_WGBS/BleachingPairs_methylseq/bismark_methylation_calls/methylation_coverage/*deduplicated.bismark.cov.gz.
merge_strands.sh (named cov_to_cyto in other lab members’ pipelines):
#!/bin/bash
#SBATCH --job-name="merge"
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --mem=500GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_WGBS/merged_cov #### this should be your new output directory so all the outputs ends up here
#SBATCH --cpus-per-task=3
#SBATCH --error="script_error_merge" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_merge" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
# load modules needed
module load Bismark/0.20.1-foss-2018b
# run coverage2cytosine merge of strands
# change paths below
# change file names below (_L003_*)
# there can't be any spaces after the \
find /data/putnamlab/estrand/BleachingPairs_WGBS/BleachingPairs_methylseq/bismark_methylation_calls/methylation_coverage/*deduplicated.bismark.cov.gz \
| xargs basename -s _L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz \
| xargs -I{} coverage2cytosine \
--genome_folder /data/putnamlab/estrand/BleachingPairs_WGBS/BleachingPairs_methylseq/reference_genome/BismarkIndex \
-o {} \
--merge_CpG \
--zero_based \
/data/putnamlab/estrand/BleachingPairs_WGBS/BleachingPairs_methylseq/bismark_methylation_calls/methylation_coverage/{}_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz
No errors in the output scripts, move on to the next step.
GENOME VERSION 3
Make a new directory for this output: mkdir merged_cov_genomev3.
merge_strands-v3.sh (named cov_to_cyto in other lab members’ pipelines):
#!/bin/bash
#SBATCH --job-name="v3-merge"
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --mem=128GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_WGBS/merged_cov_genomev3 #### this should be your new output directory so all the outputs ends up here
#SBATCH --cpus-per-task=3
#SBATCH --error="script_error_merge-v3_2" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_merge-v3_2" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
# load modules needed
module load Bismark/0.20.1-foss-2018b
# run coverage2cytosine merge of strands
# change paths below
# change file names below (_L003_*)
# there can't be any spaces after the \
find /data/putnamlab/estrand/BleachingPairs_WGBS/BleachingPairs_methylseq_v3_2/bismark_methylation_calls/methylation_coverage/*deduplicated.bismark.cov.gz \
| xargs basename -s _L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz \
| xargs -I{} coverage2cytosine \
--genome_folder /data/putnamlab/estrand/BleachingPairs_WGBS/BleachingPairs_methylseq_v3_2/reference_genome/BismarkIndex \
-o {} \
--merge_CpG \
--zero_based \
/data/putnamlab/estrand/BleachingPairs_WGBS/BleachingPairs_methylseq_v3_2/bismark_methylation_calls/methylation_coverage/{}_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz
Sanity check = ls *merged_CpG_evidence.cov | wc = 40 40 1717
Sort CpG .cov file
This function sorts all the merged files so that all scaffolds are in the same order. This needs to be done for multiIntersectBed to run correctly. This sets up a loop to do this for every sample (file).
GENOME VERSION 2
bedtools_sort.sh:
#!/bin/bash
#SBATCH --job-name="KB-sort"
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --mem=500GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_WGBS/merged_cov #### this is the output from the merge cov step above
#SBATCH --cpus-per-task=3
#SBATCH --error="script_error_sort" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_sort" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
# load BEDTools
module load BEDTools/2.27.1-foss-2018b
for f in *merged_CpG_evidence.cov
do
STEM=$(basename "${f}" .CpG_report.merged_CpG_evidence.cov)
bedtools sort -i "${f}" \
> "${STEM}"_sorted.cov
done
GENOME VERSION 3
bedtools_sort-v3.sh:
#!/bin/bash
#SBATCH --job-name="v3-KB-sort"
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --mem=128GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_WGBS/merged_cov_genomev3 #### this is the output from the merge cov step above
#SBATCH --cpus-per-task=3
#SBATCH --error="script_error_sortv3" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_sortv3" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
# load BEDTools
module load BEDTools/2.27.1-foss-2018b
for f in *merged_CpG_evidence.cov
do
STEM=$(basename "${f}" .CpG_report.merged_CpG_evidence.cov)
bedtools sort -i "${f}" \
> "${STEM}"_sorted.cov
done
Sanity check = ls *_sorted.cov | wc = 40 40 757
OVERVIEW
The script is saying:
- For every sample’s .cov file in the output folder
merged_cov, use bedtools function to sort and then output a file with the same name plus_sorted.cov.
Greate bedgraph files
GENOME VERSION 3
bedgraph-v3.sh:
#!/bin/bash
#SBATCH --job-name="bedgraph-v3"
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --mem=128GB
#SBATCH --export=NONE
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_WGBS/merged_cov_genomev3 #### this is the output from the merge cov step above
#SBATCH --cpus-per-task=3
#SBATCH --error="%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output="%x_output.%j" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
# Bedgraphs for 5X coverage
for f in *_sorted.cov
do
STEM=$(basename "${f}" _sorted.cov)
cat "${f}" | awk -F $'\t' 'BEGIN {OFS = FS} {if ($5+$6 >= 5) {print $1, $2, $3, $4}}' \
> "${STEM}"_5x_sorted.bedgraph
done
# Bedgraphs for 10X coverage
for f in *_sorted.cov
do
STEM=$(basename "${f}" _sorted.cov)
cat "${f}" | awk -F $'\t' 'BEGIN {OFS = FS} {if ($5+$6 >= 10) {print $1, $2, $3, $4}}' \
> "${STEM}"_10x_sorted.bedgraph
done
Sanity check = ls *sorted.bedgraph | wc = 80 80 2194 (x2 for two coverage levels)
Filter for a specific coverage (5X, 10X)
This script is running a loop to filter CpGs for a specified coverage and creating tab files.
Essentially, the loop in this script will take columns 5 (Methylated) and 6 (Unmethylated) positions and keeps that row if it is greater than or equal to 5. This means that we have 5x coverage for this position. This limits our interpretation to 0%, 20%, 40%, 60%, 80%, 100% methylation resolution per position.
Input File: *merged_CpG_evidence.cov
Output File: 5x_sorted.tab or 10x_sorted.tab
GENOME VERSION 2
covX.sh:
#!/bin/bash
#SBATCH --job-name="KB-5X"
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --mem=500GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_WGBS/merged_cov #### this is the output from the merge cov step above
#SBATCH --cpus-per-task=3
#SBATCH --error="script_error_5X" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_5X" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
### Filtering for CpG for 5x coverage. To change the coverage, replace X with your desired coverage in ($5+6 >= X)
for f in *_sorted.cov
do
STEM=$(basename "${f}" _sorted.cov)
cat "${f}" | awk -F $'\t' 'BEGIN {OFS = FS} {if ($5+$6 >= 5) {print $1, $2, $3, $4, $5, $6}}' \
> "${STEM}"_5x_sorted.tab
done
### Filtering for CpG for 10x coverage. To change the coverage, replace X with your desired coverage in ($5+6 >= X)
for f in *_sorted.cov
do
STEM=$(basename "${f}" _sorted.cov)
cat "${f}" | awk -F $'\t' 'BEGIN {OFS = FS} {if ($5+$6 >= 10) {print $1, $2, $3, $4, $5, $6}}' \
> "${STEM}"_10x_sorted.tab
done
GENOME VERSION 3
covX-v3.sh:
#!/bin/bash
#SBATCH --job-name="v3-KB-5X"
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --mem=128GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_WGBS/merged_cov_genomev3 #### this is the output from the merge cov step above
#SBATCH --cpus-per-task=3
#SBATCH --error="script_error_5X-v3" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_5X-v3" #once your job is completed, any final job report comments will be put in this file
# load modules needed (specific need for my computer)
source /usr/share/Modules/init/sh # load the module function
### Filtering for CpG for 5x coverage. To change the coverage, replace X with your desired coverage in ($5+6 >= X)
for f in *_sorted.cov
do
STEM=$(basename "${f}" _sorted.cov)
cat "${f}" | awk -F $'\t' 'BEGIN {OFS = FS} {if ($5+$6 >= 5) {print $1, $2, $3, $4, $5, $6}}' \
> "${STEM}"_5x_sorted.tab
done
### Filtering for CpG for 10x coverage. To change the coverage, replace X with your desired coverage in ($5+6 >= X)
for f in *_sorted.cov
do
STEM=$(basename "${f}" _sorted.cov)
cat "${f}" | awk -F $'\t' 'BEGIN {OFS = FS} {if ($5+$6 >= 10) {print $1, $2, $3, $4, $5, $6}}' \
> "${STEM}"_10x_sorted.tab
done
Sanity check ls *sorted.tab | wc = 80 80 1794 (x2 for two coverage levels)
OVERVIEW
Moving forward I want to see the differences in data we get from 5X and 10X. We’ll have to decide which threshold to use moving forward. We want confidence and high resolution but also a large dataset so we need a happy medium.
5x coverage
wc -l *5x_sorted.tab:
10795712 16_S138_5x_sorted.tab
9475520 17_S134_5x_sorted.tab
10544006 18_S154_5x_sorted.tab
11086014 21_S119_5x_sorted.tab
8324187 22_S120_5x_sorted.tab
9565847 23_S141_5x_sorted.tab
7217442 24_S147_5x_sorted.tab
15508592 25_S148_5x_sorted.tab
6679878 26_S121_5x_sorted.tab
13129157 28_S122_5x_sorted.tab
13177726 29_S153_5x_sorted.tab
11203815 2_S128_5x_sorted.tab
10223642 30_S124_5x_sorted.tab
1414587 31_S127_5x_sorted.tab
10908998 32_S130_5x_sorted.tab
7641308 33_S142_5x_sorted.tab
9390715 34_S136_5x_sorted.tab
12071740 35_S137_5x_sorted.tab
8944977 37_S140_5x_sorted.tab
6851807 38_S129_5x_sorted.tab
10867493 39_S145_5x_sorted.tab
11144799 40_S135_5x_sorted.tab
8654965 41_S151_5x_sorted.tab
10887332 42_S131_5x_sorted.tab
12484052 43_S143_5x_sorted.tab
9865682 44_S125_5x_sorted.tab
8637091 45_S156_5x_sorted.tab
8379720 46_S133_5x_sorted.tab
10301106 47_S155_5x_sorted.tab
14300207 4_S146_5x_sorted.tab
11738047 50_S139_5x_sorted.tab
8229716 51_S126_5x_sorted.tab
7771771 52_S150_5x_sorted.tab
11123515 54_S144_5x_sorted.tab
7765416 55_S123_5x_sorted.tab
6517952 56_S149_5x_sorted.tab
6887246 57_S152_5x_sorted.tab
6034212 58_S157_5x_sorted.tab
6793494 59_S158_5x_sorted.tab
11607615 6_S132_5x_sorted.tab
384147101 total
10x coverage
wc -l *10x_sorted.tab:
2849086 16_S138_10x_sorted.tab
1898361 17_S134_10x_sorted.tab
2928908 18_S154_10x_sorted.tab
3191365 21_S119_10x_sorted.tab
1454107 22_S120_10x_sorted.tab
1885121 23_S141_10x_sorted.tab
1044970 24_S147_10x_sorted.tab
9192668 25_S148_10x_sorted.tab
839032 26_S121_10x_sorted.tab
5449797 28_S122_10x_sorted.tab
5380052 29_S153_10x_sorted.tab
3214753 2_S128_10x_sorted.tab
2522178 30_S124_10x_sorted.tab
40842 31_S127_10x_sorted.tab
2921673 32_S130_10x_sorted.tab
1044145 33_S142_10x_sorted.tab
1893638 34_S136_10x_sorted.tab
3994034 35_S137_10x_sorted.tab
1587468 37_S140_10x_sorted.tab
800425 38_S129_10x_sorted.tab
3126530 39_S145_10x_sorted.tab
3254802 40_S135_10x_sorted.tab
1972497 41_S151_10x_sorted.tab
3235614 42_S131_10x_sorted.tab
4763284 43_S143_10x_sorted.tab
2320334 44_S125_10x_sorted.tab
1724432 45_S156_10x_sorted.tab
1469327 46_S133_10x_sorted.tab
2746614 47_S155_10x_sorted.tab
6886932 4_S146_10x_sorted.tab
3872754 50_S139_10x_sorted.tab
1505077 51_S126_10x_sorted.tab
1401552 52_S150_10x_sorted.tab
3448169 54_S144_10x_sorted.tab
1282952 55_S123_10x_sorted.tab
851327 56_S149_10x_sorted.tab
961452 57_S152_10x_sorted.tab
893503 58_S157_10x_sorted.tab
944123 59_S158_10x_sorted.tab
3462425 6_S132_10x_sorted.tab
104256323 total
Create a file with positions found in all samples
We need to create a file that is filtered to only positions that are found in all samples (both methylated and unmethylated). multiIntersectBed creates a file that merges all samples together. The 4th column then tells you how samples have that position. We can then filter positions based on this column that is equal to our sample size. n=40 for this project.
Input file: 5x_sorted.tab and 10x_sorted.tab
Output file: CpG.filt.all.samps.5x_sorted.bed and CpG.filt.all.samps.10x_sorted.bed
GENOME VERSION 3
cov_allsamples-v3.sh:
#!/bin/bash
#SBATCH --job-name="v3-KB-all_cov"
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --mem=128GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_WGBS/merged_cov_genomev3 #### this is the output from the merge cov step above
#SBATCH --cpus-per-task=3
#SBATCH --error="script_error_all_cov-v3" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_all_cov-v3" #once your job is completed, any final job report comments will be put in this file
# load modules needed
source /usr/share/Modules/init/sh # load the module function (specific to my computer)
module load BEDTools/2.27.1-foss-2018b
multiIntersectBed -i *_5x_sorted.tab > CpG.all.samps.5x_sorted.bed
multiIntersectBed -i *_10x_sorted.tab > CpG.all.samps.10x_sorted.bed
cat CpG.all.samps.5x_sorted.bed | awk '$4 ==40' > CpG.filt.all.samps.5x_sorted.bed
cat CpG.all.samps.10x_sorted.bed | awk '$4 ==40' > CpG.filt.all.samps.10x_sorted.bed
head -2 CpG.filt.all.samps.10x_sorted.bed:
Montipora_capitata_HIv3___Scaffold_1 15259 15261 1 2 0 1 0 0 0 0 0 0 0 0 0 0 00 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 00 0 0 0 0
Montipora_capitata_HIv3___Scaffold_1 15275 15277 1 2 0 1 0 0 0 0 0 0 0 0 0 0 00 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 00 0 0 0 0
head -2 CpG.filt.all.samps.5x_sorted.bed:
Montipora_capitata_HIv3___Scaffold_1 25587 25589 40 1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 11 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
Montipora_capitata_HIv3___Scaffold_1 25599 25601 40 1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 11 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
wc -l CpG.filt.all.samps.5x_sorted.bed:
96449 CpG.filt.all.samps.5x_sorted.bed
wc -l CpG.filt.all.samps.10x_sorted.bed:
4512 CpG.filt.all.samps.10x_sorted.bed
Gene annotation
This step needs a modified gff file that is only includes gene positions. This can be found on the Rutger’s data site for M. capiata genome resources: http://cyanophora.rutgers.edu/montipora/. I used v2 of the genome previously in this script and then a more updated version of the genome came out (v3). I re-ran all above steps with v3 genome and will be using only v3 from this point on.
$ wget http://cyanophora.rutgers.edu/montipora/Montipora_capitata_HIv3.genes.gff3.gz and gunzip Montipora_capitata_HIv3.genes.gff3.gz.
File name = Montipora_capitata_HIv3.genes.gff3:
Montipora_capitata_HIv3___Scaffold_12 AUGUSTUS transcript 27295 28641 . - . ID=Montipora_capitata_HIv3___RNAseq.g37162.t1
Montipora_capitata_HIv3___Scaffold_12 AUGUSTUS CDS 27295 28641 . - 0 Parent=Montipora_capitata_HIv3___RNAseq.g37162.t1
Montipora_capitata_HIv3___Scaffold_12 AUGUSTUS exon 27295 28641 . - 0 Parent=Montipora_capitata_HIv3___RNAseq.g37162.t1
Montipora_capitata_HIv3___Scaffold_12 AUGUSTUS transcript 40222 40725 . - . ID=Montipora_capitata_HIv3___TS.g29675.t1
Montipora_capitata_HIv3___Scaffold_12 AUGUSTUS CDS 40222 40725 . - 0 Parent=Montipora_capitata_HIv3___TS.g29675.t1
Montipora_capitata_HIv3___Scaffold_12 AUGUSTUS exon 40222 40725 . - 0 Parent=Montipora_capitata_HIv3___TS.g29675.t1
Filtering the 3rd column for only ‘genes’:
$ awk '{if ($3 == "gene") {print}}' Montipora_capitata_HIv3.genes.gff3 > Montipora_capitata_HIv3.genes_only.gff3
This came back empty so the descriptions are all parts of genes. Removing the created file above and moving on with the original file (Montipora_capitata_HIv3.genes.gff3). (In other pipelines our lab has done, we needed this step).
Do I need to filter this to transcript instead?
IntersectBed: Loci mapped to annotated gene
Next, merge each sample file with gene annotation file using intersectBed.
Input files: *5x_sorted.tab and *10x_sorted.tab and Montipora_capitata_HIv3.genes.gff3
Output files: *_5x_sorted.tab_gene and *_10x_sorted.tab_gene
GENOME VERSION 3
intersectBed-v3.sh:
#!/bin/bash
#SBATCH --job-name="v3KB-intersectBed"
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --mem=128GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_WGBS/merged_cov_genomev3 #### this is the output from the merge cov step above
#SBATCH --cpus-per-task=3
#SBATCH --error="script_error_intersectBed-v3" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_intersectBed-v3" #once your job is completed, any final job report comments will be put in this file
# load modules needed
source /usr/share/Modules/init/sh # load the module function (specific to my computer)
module load BEDTools/2.27.1-foss-2018b
for i in *5x_sorted.tab
do
intersectBed \
-wb \
-a ${i} \
-b /data/putnamlab/estrand/Montipora_capitata_HIv3.genes.gff3 \
> ${i}_gene
done
for i in *10x_sorted.tab
do
intersectBed \
-wb \
-a ${i} \
-b /data/putnamlab/estrand/Montipora_capitata_HIv3.genes.gff3 \
> ${i}_gene
done
Sanity check ls *_gene | wc = 80 80 2194
The above code probably doesn’t need to be divided into 5 and 10x if the function is the same and the names of the files have 5 or 10 already in them.
IntersectBed: File to only positions found in all samples
GENOME VERSION 3
intersect_enrichment-v3.sh
#!/bin/bash
#SBATCH --job-name="v3KB-enrich"
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --mem=128GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_WGBS/merged_cov_genomev3 #### this is the output from the merge cov step above
#SBATCH --cpus-per-task=3
#SBATCH --error="script_error_v3KB-enrich" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_v3KB-enrich" #once your job is completed, any final job report comments will be put in this file
# load modules needed
source /usr/share/Modules/init/sh # load the module function (specific to my computer)
module load BEDTools/2.27.1-foss-2018b
for i in *_5x_sorted.tab_gene
do
intersectBed \
-a ${i} \
-b CpG.filt.all.samps.5x_sorted.bed \
> ${i}_CpG_5x_enrichment.bed
done
for i in *_10x_sorted.tab_gene
do
intersectBed \
-a ${i} \
-b CpG.filt.all.samps.10x_sorted.bed \
> ${i}_CpG_10x_enrichment.bed
done
ls *CpG_10x_enrichment.bed | wc = 40
ls *CpG_5x_enrichment.bed | wc = 40
Within merged_cov_genomev3 folder:
wc -l *10x_enrichment.bed > 10x_enrichment_sample_size.txt
wc -l *5x_enrichment.bed > 5x_enrichment_sample_size.txt
5X COVERAGE
87180 16_S138_5x_sorted.tab_gene_CpG_5x_enrichment.bed
87180 17_S134_5x_sorted.tab_gene_CpG_5x_enrichment.bed
87180 18_S154_5x_sorted.tab_gene_CpG_5x_enrichment.bed
87180 21_S119_5x_sorted.tab_gene_CpG_5x_enrichment.bed
87180 22_S120_5x_sorted.tab_gene_CpG_5x_enrichment.bed
87180 23_S141_5x_sorted.tab_gene_CpG_5x_enrichment.bed
87180 24_S147_5x_sorted.tab_gene_CpG_5x_enrichment.bed
87180 25_S148_5x_sorted.tab_gene_CpG_5x_enrichment.bed
87180 26_S121_5x_sorted.tab_gene_CpG_5x_enrichment.bed
87180 28_S122_5x_sorted.tab_gene_CpG_5x_enrichment.bed
10X COVERAGE
1667 16_S138_10x_sorted.tab_gene_CpG_10x_enrichment.bed
1667 17_S134_10x_sorted.tab_gene_CpG_10x_enrichment.bed
1667 18_S154_10x_sorted.tab_gene_CpG_10x_enrichment.bed
1667 21_S119_10x_sorted.tab_gene_CpG_10x_enrichment.bed
1667 22_S120_10x_sorted.tab_gene_CpG_10x_enrichment.bed
1667 23_S141_10x_sorted.tab_gene_CpG_10x_enrichment.bed
1667 24_S147_10x_sorted.tab_gene_CpG_10x_enrichment.bed
1667 25_S148_10x_sorted.tab_gene_CpG_10x_enrichment.bed
1667 26_S121_10x_sorted.tab_gene_CpG_10x_enrichment.bed
1667 28_S122_10x_sorted.tab_gene_CpG_10x_enrichment.bed
Export Files
scp 'emma_strand@ssh3.hac.uri.edu:/data/putnamlab/estrand/BleachingPairs_WGBS/merged_cov_genomev3/*_5x_enrichment.bed' ~/MyProjects/HI_Bleaching_Timeseries/Dec-July-2019-analysis/output/WGBS/meth_counts_5x/
scp 'emma_strand@ssh3.hac.uri.edu:/data/putnamlab/estrand/BleachingPairs_WGBS/merged_cov_genomev3/*_10x_enrichment.bed' ~/MyProjects/HI_Bleaching_Timeseries/Dec-July-2019-analysis/output/WGBS/meth_counts_10x
Troubleshooting
The files that did work the first time (in BleachingPairs_methylseq_v3):
17_S134_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz 38_S129_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz
18_S154_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz 39_S145_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz
21_S119_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz 40_S135_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz
22_S120_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz 42_S131_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz
23_S141_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz 43_S143_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz
24_S147_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz 44_S125_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz
25_S148_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz 45_S156_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz
26_S121_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz 4_S146_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz
28_S122_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz 50_S139_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz
2_S128_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz 54_S144_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz
31_S127_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz 58_S157_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz
33_S142_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz 59_S158_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz
34_S136_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz 6_S132_L003_R1_001_val_1_bismark_bt2_pe.deduplicated.bismark.cov.gz
Files that did not run the first time: 16, 29, 30, 32, 35, 37, 41, 46, 47, 51, 52, 55, 56, 57
but the step before worked b/c the output less 16_S138_L003_R1_001_val_1_bismark_bt2_pe.deduplication_report.txt yields:
Total number of alignments analysed in 16_S138_L003_R1_001_val_1_bismark_bt2_pe.bam: 23895598
Total number duplicated alignments removed: 2940163 (12.30%)
Duplicated alignments were found at: 2599109 different position(s)
Total count of deduplicated leftover sequences: 20955435 (87.70% of total)
So why would this not get used as input for methylation calling….
| cat CpG.all.samps.5x_sorted.bed | awk ‘$5 ==”,40”’ > CpG.all.samps-40.5x_sorted.bed (awk contains samples 26-40) to make sure all samples do exist in all samples file |
list all sample names manually to make sure all 40 are
head CpG.all.samps.5x_sorted.bed:
Montipora_capitata_HIv3___Scaffold_1 15160 15162 2 1,16 1 0 0 0 0 0 0 0 0 0 0 0
0 0 0 1 0 0 0 0 0 0 0 0 0 0
Montipora_capitata_HIv3___Scaffold_1 15182 15184 2 1,16 1 0 0 0 0 0 0 0 0 0 0 0
0 0 0 1 0 0 0 0 0 0 0 0 0 0
Montipora_capitata_HIv3___Scaffold_1 15206 15210 2 1,16 1 0 0 0 0 0 0 0 0 0 0 0
0 0 0 1 0 0 0 0 0 0 0 0 0 0
Montipora_capitata_HIv3___Scaffold_1 15226 15228 2 1,16 1 0 0 0 0 0 0 0 0 0 0 0
0 0 0 1 0 0 0 0 0 0 0 0 0 0
Montipora_capitata_HIv3___Scaffold_1 15259 15261 11 1,3,7,10,12,13,16,17,19,22,23 1 0 1 0 0 0 1 0 0
1 0 1 1 0 0 1 1 0 1 0 0 1 1 0 0 0
Montipora_capitata_HIv3___Scaffold_1 15275 15277 10 1,3,4,7,10,12,13,16,19,22 1 0 1 1 0 0 1 0 0
1 0 1 1 0 0 1 0 0 1 0 0 1 0 0 0 0
head CpG.filt.all.samps.5x_sorted.bed:
empty..
Try with lower # of column #4 value – this cuts off after loci found in 26 samples or more.
cat CpG.all.samps.5x_sorted.bed | awk '$4 ==26' > CpG.filt.all.samps.5x_sorted-26.bed
wc -l CpG.filt.all.samps.5x_sorted-26.bed
739760 CpG.filt.all.samps.5x_sorted-10.bed
776113 CpG.filt.all.samps.5x_sorted-15.bed
420987 CpG.filt.all.samps.5x_sorted-25.bed
158992 CpG.filt.all.samps.5x_sorted-26.bed
0 CpG.filt.all.samps.5x_sorted-27.bed
0 CpG.filt.all.samps.5x_sorted-30.bed