
KBay Bleaching Pairs RNASeq Pipeline Analysis
KBay Bleaching Pairs RNASeq Pipeline Analysis
Contents:
- Project Details
- Setting Up Andromeda
- FastQC
- Trim Reads
- Align Trimmed Reads to Reference Genome
- Assemble aligned reads and quantify transcripts
- Create Gene Counts Matrix
KBay Bleaching Pairs project details
Project github: HI_Bleaching_Pairs
Molecular laboratory work spreadsheet: excel doc
40 adult coral biopsies of M. capitata used for molecular analysis from July 2019 and December 2019 time points. Laboratory work for this project found here.
Raw data path: ../../data/putnamlab/KITT/hputnam/20220203_BleachedPairs_RNASeq
Sym linked files in my working directory: ../../data/putnamlab/estrand/BleachingPairs_RNASeq
Script to download raw data found here in github repo.
A. Huffmyer first-pass analysis of these files: /data/putnamlab/ashuffmyer/pairs-rnaseq (done with Version 2 of genome)
M. capitata genome database: http://cyanophora.rutgers.edu/montipora/. I will use version 3 for this analysis.
References:
For detailed explanations of each step, refer to the below pipelines.
RNASeq
- Erin Chille RNAseq pipeline: https://github.com/echille/Mcapitata_Developmental_Gene_Expression_Timeseries/blob/master/1-QC-Align-Assemble/mcap_rnaseq_analysis.md
- Jill Ashey RNAseq pipeline: https://github.com/JillAshey/SedimentStress/blob/master/Bioinf/RNASeq_pipeline_HI.md
- Danielle Becker pipeline: https://github.com/daniellembecker/DanielleBecker_Lab_Notebook/blob/master/_posts/2021-04-14-Molecular-Underpinnings-RNAseq-Workflow.md
TagSeq
- Kevin Wong pipeline: https://github.com/kevinhwong1/Porites_Rim_Bleaching_2019/blob/master/scripts/TagSeq/TagSeq_Analysis_HPC.md
Function Annotation pipeline
See here for M. capitata genome version 3 functional annotation: https://github.com/emmastrand/EmmaStrand_Notebook/blob/master/_posts/2022-11-02-M.capitata-Genome-v3-Functional-Annotation.md.
Setting Up Andromeda
Sym link raw files (from HP path above) to raw_data folder within /BleachingPairs_RNASeq.
Make a new directory for scripts, fastqc_results
FastQC
fastqc.sh:
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=emma_strand@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq
#SBATCH --error="script_error_fastqc" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_fastqc" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function (specifc to my computer)
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
for file in /data/putnamlab/KITT/hputnam/20220203_BleachedPairs_RNASeq/*fastq.gz
do
fastqc $file --outdir /data/putnamlab/estrand/BleachingPairs_RNASeq/fastqc_results/
done
multiqc --interactive /data/putnamlab/estrand/BleachingPairs_RNASeq/fastqc_results -o /data/putnamlab/estrand/BleachingPairs_RNASeq/multiqc_results
Outside of Andromeda:
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/BleachingPairs_RNASeq/multiqc_results/multiqc_report.html /Users/emmastrand/MyProjects/HI_Bleaching_Timeseries/Dec-July-2019-analysis/output/RNASeq/initial_multiqc_report.html
fastqc results
All samples have sequences of a single length (101bp).
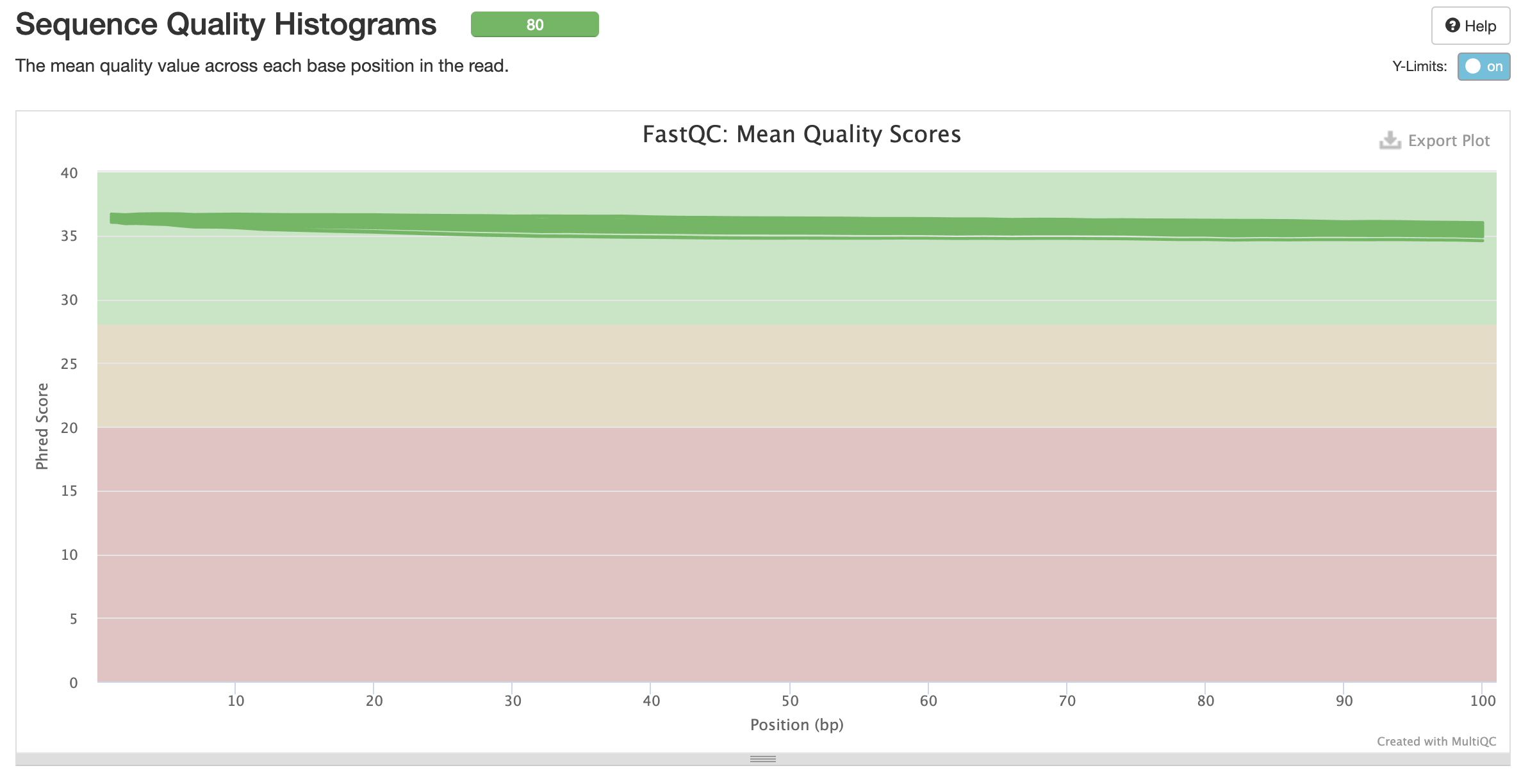
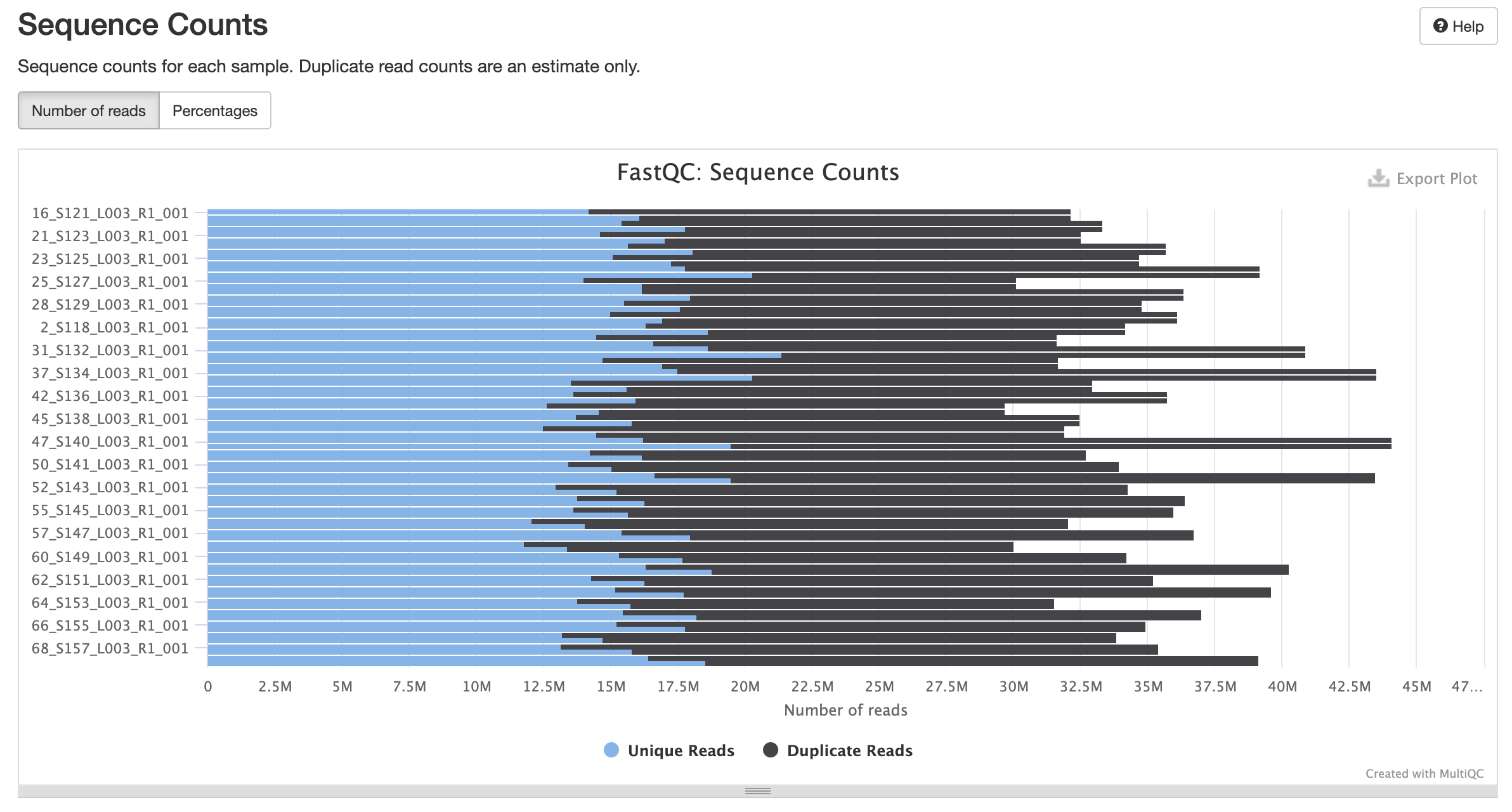




Trimming Reads
I use the program fastp, but there are others like Trimmomatic.
- More info on Trimmomatic from Data Carpentry.
- More info on fastp https://manpages.debian.org/testing/fastp/fastp.1.en.html
fastp_multiqc.sh:
Fill in the trimming parameters based on the initial multiqc report.
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/raw_data
#SBATCH --error=../"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function (specific to my computer)
# Load modules needed
module load fastp/0.19.7-foss-2018b
module load FastQC/0.11.8-Java-1.8
module load MultiQC/1.9-intel-2020a-Python-3.8.2
# Make an array (list) of sequences to trim
# Needs to be in directory above (raw data directory)
array1=($(ls *.fastq.gz))
# fastp and fastqc loop
for i in ${array1[@]}; do
fastp --in1 ${i} \
--in2 $(echo ${i}|sed s/_R1/_R2/)\
--out1 /data/putnamlab/estrand/BleachingPairs_RNASeq/processed/trimmed.${i} \
--out2 /data/putnamlab/estrand/BleachingPairs_RNASeq/processed/trimmed.$(echo ${i}|sed s/_R1/_R2/) \
--qualified_quality_phred 20 \
--unqualified_percent_limit 10 \
--length_required 100 \
--detect_adapter_for_pe \
--cut_right cut_right_window_size 5 cut_right_mean_quality 20
fastqc /data/putnamlab/estrand/BleachingPairs_RNASeq/processed/trimmed.${i}
done
echo "Read trimming of adapters complete." $(date)
# Quality Assessment of Trimmed Reads
cd /data/putnamlab/estrand/BleachingPairs_RNASeq/processed/ #The following command will be run in the /clean directory
# Compile MultiQC report from FastQC files
multiqc --interactive ./
echo "Cleaned MultiQC report generated." $(date)
Exporting this report (outside of Andromeda)
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/BleachingPairs_RNASeq/processed/multiqc_report.html /Users/emmastrand/MyProjects/HI_Bleaching_Timeseries/Dec-July-2019-analysis/output/RNASeq/processed_multiqc_report.html
Processed Multiqc report
80 samples had less than 1% of reads made up of overrepresented sequences
No samples found with any adapter contamination > 0.1%
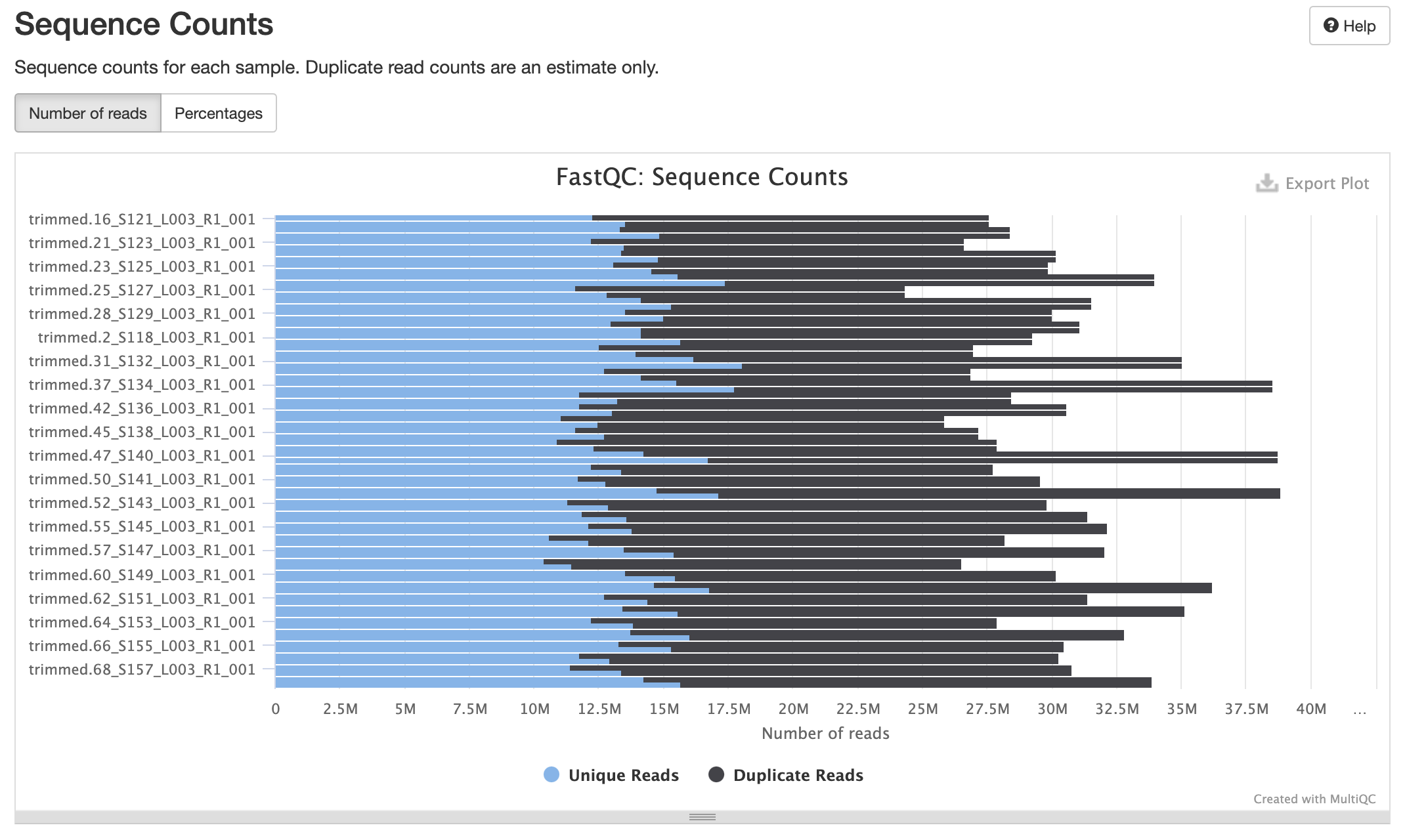





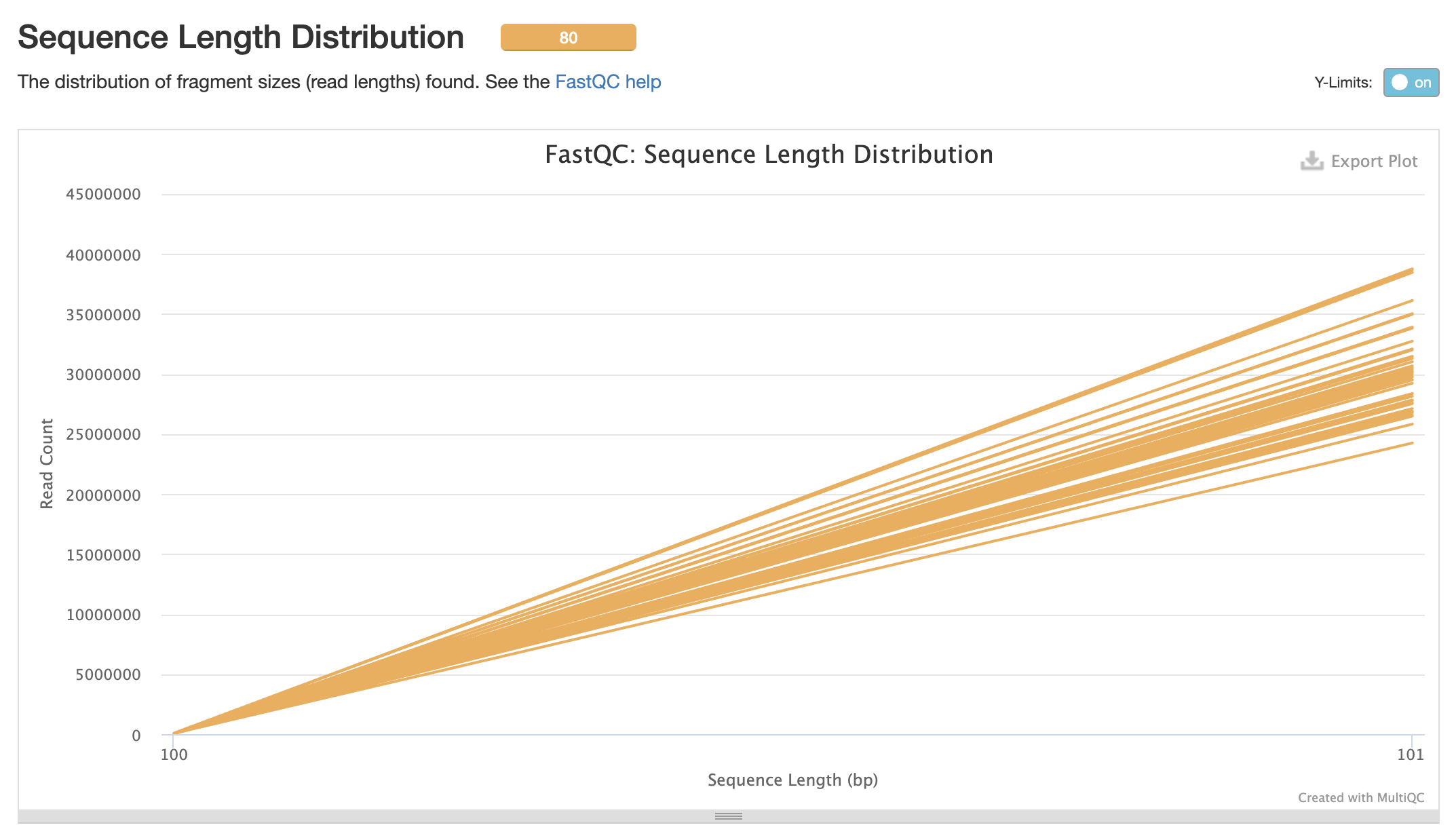
Align Trimmed Reads to Reference Genome
I use the program HISAT2, but other pipelines use STAR.
- Index the reference genome
- Alignment of clean reads to the reference genome
Reference genome information
I’m using the newest version of the Mcap genome:
wget http://cyanophora.rutgers.edu/montipora/Montipora_capitata_HIv3.assembly.fasta.gz. gunzip this file
wget http://cyanophora.rutgers.edu/montipora/Montipora_capitata_HIv3.genes.gff3.gz. gunzip this file
path: /data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta
path: /data/putnamlab/estrand/Montipora_capitata_HIv3.genes.gff3
Creating the reference genome part takes ~5-10 minutes.
Running the hisat2 and samtools parts took under 4 days (the align.sh script at the end of this doc took 2 weeks..)
align2.sh:
#!/bin/bash
#SBATCH -t 500:00:00
#SBATCH --export=NONE
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/output2/
#SBATCH --error=../"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function (specific need to my computer)
# load modules needed
module load HISAT2/2.2.1-gompi-2021b
module load SAMtools/1.15.1-GCC-11.2.0
# Index the reference genome
hisat2-build -f /data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta ./Mcap_ref
echo "Reference genome indexed. Starting alignment" $(date)
# Alignment of clean reads to the reference genome
array=($(ls /data/putnamlab/estrand/BleachingPairs_RNASeq/processed/*_R1_001.fastq.gz))
for i in ${array[@]}; do
hisat2 -p 8 --rna-strandness RF --dta -q -x Mcap_ref -1 ${i} -2 $(echo ${i}|sed s/_R1/_R2/) -S ${i}.sam
samtools sort -@ 8 -o ${i}.bam ${i}.sam
echo "${i} bam-ified!"
rm ${i}.sam
done
OUTPUT SHOULD BE BOTH R1 AND R2 IN ONE SAM FILE!!
Assemble aligned reads and quantify transcripts
StringTie is a fast and highly efficient assembler of RNA-Seq alignments into potential transcripts.
- Reference-guided assembly with novel transcript discovery
- Merge output GTF files and assess the assembly performance
- Compilation of GTF-files into gene and transcript count matrices
GFF File Fix
My original counts matrix output with names like STRG.6173 instead of the gene name.
Ariana had a solution to this outlined here: https://github.com/Putnam-Lab/Lab_Management/issues/51
Upload new GFF file
scp /Users/emmastrand/MyProjects/HI_Bleaching_Timeseries/Dec-July-2019-analysis/Montipora_capitata_HIv3.genes_fixed.gff3.gz emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/
Ended up using assemble2.sh below (b/c of a formatting issue in my original gff)
assemble2.sh:
#!/bin/bash
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=128GB
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/output2/
#SBATCH --error=../"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function (specific need to my computer)
module load StringTie/2.1.4-GCC-9.3.0
# Assemble and estimate reads
# array 1 ls is every file that ends with .bam in the output2 folder specified above
array1=($(ls *.bam))
for i in ${array1[@]}; do
stringtie -p 8 -e -B -G /data/putnamlab/estrand/Montipora_capitata_HIv3.genes_fixed.gff3 -o /data/putnamlab/estrand/BleachingPairs_RNASeq/output2/${i}.gtf /data/putnamlab/estrand/BleachingPairs_RNASeq/output2/${i}
done
# Merge stringTie gtf results
ls trimmed.*.gtf > mergelist.txt
cat mergelist.txt
stringtie --merge -p 8 -G /data/putnamlab/estrand/Montipora_capitata_HIv3.genes_fixed.gff3 -o stringtie_merged.gtf mergelist.txt
echo "Stringtie merge complete" $(date)
head mergelist.txt:
trimmed.16_S121_L003_R1_001.fastq.gz.bam.gtf
trimmed.17_S122_L003_R1_001.fastq.gz.bam.gtf
trimmed.21_S123_L003_R1_001.fastq.gz.bam.gtf
trimmed.22_S124_L003_R1_001.fastq.gz.bam.gtf
trimmed.23_S125_L003_R1_001.fastq.gz.bam.gtf
trimmed.24_S126_L003_R1_001.fastq.gz.bam.gtf
trimmed.25_S127_L003_R1_001.fastq.gz.bam.gtf
trimmed.26_S128_L003_R1_001.fastq.gz.bam.gtf
trimmed.28_S129_L003_R1_001.fastq.gz.bam.gtf
head stringtie_merged.gtf:
# stringtie --merge -p 8 -G /data/putnamlab/estrand/Montipora_capitata_HIv3.genes_fixed.gff3 -o stringtie_merged.gtf mergelist.txt
# StringTie version 2.1.4
Montipora_capitata_HIv3___Scaffold_1 StringTie transcript 22732 36176 1000 - . gene_id "MSTRG.1"; transcript_id "Montipora_capitata_HIv3___RNAseq.g4581.t1"; ref_gene_id "Montipora_capitata_HIv3___RNAseq.g4581.t1";
Montipora_capitata_HIv3___Scaffold_1 StringTie exon 22732 23002 1000 - . gene_id "MSTRG.1"; transcript_id "Montipora_capitata_HIv3___RNAseq.g4581.t1"; exon_number "1"; ref_gene_id "Montipora_capitata_HIv3___RNAseq.g4581.t1";
Montipora_capitata_HIv3___Scaffold_1 StringTie exon 27868 27934 1000 - . gene_id "MSTRG.1"; transcript_id "Montipora_capitata_HIv3___RNAseq.g4581.t1"; exon_number "2"; ref_gene_id "Montipora_capitata_HIv3___RNAseq.g4581.t1";
Montipora_capitata_HIv3___Scaffold_1 StringTie exon 29683 29722 1000 - . gene_id "MSTRG.1"; transcript_id "Montipora_capitata_HIv3___RNAseq.g4581.t1"; exon_number "3"; ref_gene_id "Montipora_capitata_HIv3___RNAseq.g4581.t1";
Montipora_capitata_HIv3___Scaffold_1 StringTie exon 30150 30230 1000 - . gene_id "MSTRG.1"; transcript_id "Montipora_capitata_HIv3___RNAseq.g4581.t1"; exon_number "4"; ref_gene_id "Montipora_capitata_HIv3___RNAseq.g4581.t1";
Montipora_capitata_HIv3___Scaffold_1 StringTie exon 31466 31705 1000 - . gene_id "MSTRG.1"; transcript_id "Montipora_capitata_HIv3___RNAseq.g4581.t1"; exon_number "5"; ref_gene_id "Montipora_capitata_HIv3___RNAseq.g4581.t1";
Montipora_capitata_HIv3___Scaffold_1 StringTie exon 31890 31988 1000 - . gene_id "MSTRG.1"; transcript_id "Montipora_capitata_HIv3___RNAseq.g4581.t1"; exon_number "6"; ref_gene_id "Montipora_capitata_HIv3___RNAseq.g4581.t1";
Montipora_capitata_HIv3___Scaffold_1 StringTie exon 33295 33367 1000 - . gene_id "MSTRG.1"; transcript_id "Montipora_capitata_HIv3___RNAseq.g4581.t1"; exon_number "7"; ref_gene_id "Montipora_capitata_HIv3___RNAseq.g4581.t1";
Create gene counts matrix
The StringTie program includes a script, prepDE.py that compiles your assembly files into gene and transcript count matrices. This script requires as input the list of sample names and their full file paths, sample_list.txt. This file will live in StringTie program directory.
Copy this file: cp /data/putnamlab/ashuffmyer/pairs-rnaseq/prepDE.py .
Ended up using genecount2.sh b/c of a formatting issue above
genecount2.sh:
#!/bin/bash
#SBATCH -t 60:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=128GB
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/output2/
#SBATCH --error=../"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function (specific need to my computer)
module load StringTie/2.1.4-GCC-9.3.0
module load Python/2.7.15-foss-2018b
module load GffCompare/0.12.1-GCCcore-8.3.0
# Assess the performance of the assembly
gffcompare -r /data/putnamlab/estrand/Montipora_capitata_HIv3.genes_fixed.gff3 -o merged stringtie_merged.gtf
echo "GFFcompare complete, Starting gene count matrix assembly..." $(date)
# Make gtf list text file
for filename in *.bam.gtf; do echo $filename $PWD/$filename; done > listGTF.txt
# Compile the gene count matrix
python ../scripts/prepDE.py -g ../KB_gene_count_matrix2.csv -i listGTF.txt
echo "Gene count matrix compiled." $(date)
head output2/listGTF.txt :
trimmed.16_S121_L003_R1_001.fastq.gz.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output2/trimmed.16_S121_L003_R1_001.fastq.gz.bam.gtf
trimmed.17_S122_L003_R1_001.fastq.gz.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output2/trimmed.17_S122_L003_R1_001.fastq.gz.bam.gtf
trimmed.21_S123_L003_R1_001.fastq.gz.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output2/trimmed.21_S123_L003_R1_001.fastq.gz.bam.gtf
trimmed.22_S124_L003_R1_001.fastq.gz.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output2/trimmed.22_S124_L003_R1_001.fastq.gz.bam.gtf
trimmed.23_S125_L003_R1_001.fastq.gz.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output2/trimmed.23_S125_L003_R1_001.fastq.gz.bam.gtf
trimmed.24_S126_L003_R1_001.fastq.gz.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output2/trimmed.24_S126_L003_R1_001.fastq.gz.bam.gtf
trimmed.25_S127_L003_R1_001.fastq.gz.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output2/trimmed.25_S127_L003_R1_001.fastq.gz.bam.gtf
trimmed.26_S128_L003_R1_001.fastq.gz.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output2/trimmed.26_S128_L003_R1_001.fastq.gz.bam.gtf
trimmed.28_S129_L003_R1_001.fastq.gz.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output2/trimmed.28_S129_L003_R1_001.fastq.gz.bam.gtf
trimmed.29_S130_L003_R1_001.fastq.gz.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output2/trimmed.29_S130_L003_R1_001.fastq.gz.bam.gtf
head KB_gene_count_matrix.csv:
gene_id,trimmed.16_S121_L003_R1_001.fastq.gz.bam.gtf,trimmed.17_S122_L003_R1_001.fastq.gz.bam.gtf,trimmed.21_S123_L003_R1_001.fastq.gz.bam.gtf,trimmed.22_S124_L003_R1_001.fastq.gz.bam.gtf,trimmed.23_S125_L003_R1_001.fastq.gz.bam.gtf,trimmed.24_S126_L003_R1_001.fastq.gz.bam.gtf,trimmed.25_S127_L003_R1_001.fastq.gz.bam.gtf,trimmed.26_S128_L003_R1_001.fastq.gz.bam.gtf,trimmed.28_S129_L003_R1_001.fastq.gz.bam.gtf,trimmed.29_S130_L003_R1_001.fastq.gz.bam.gtf,trimmed.2_S118_L003_R1_001.fastq.gz.bam.gtf,trimmed.30_S131_L003_R1_001.fastq.gz.bam.gtf,trimmed.31_S132_L003_R1_001.fastq.gz.bam.gtf,trimmed.33_S133_L003_R1_001.fastq.gz.bam.gtf,trimmed.37_S134_L003_R1_001.fastq.gz.bam.gtf,trimmed.39_S135_L003_R1_001.fastq.gz.bam.gtf,trimmed.42_S136_L003_R1_001.fastq.gz.bam.gtf,trimmed.43_S137_L003_R1_001.fastq.gz.bam.gtf,trimmed.45_S138_L003_R1_001.fastq.gz.bam.gtf,trimmed.46_S139_L003_R1_001.fastq.gz.bam.gtf,trimmed.47_S140_L003_R1_001.fastq.gz.bam.gtf,trimmed.4_S119_L003_R1_001.fastq.gz.bam.gtf,trimmed.50_S141_L003_R1_001.fastq.gz.bam.gtf,trimmed.51_S142_L003_R1_001.fastq.gz.bam.gtf,trimmed.52_S143_L003_R1_001.fastq.gz.bam.gtf,trimmed.54_S144_L003_R1_001.fastq.gz.bam.gtf,trimmed.55_S145_L003_R1_001.fastq.gz.bam.gtf,trimmed.56_S146_L003_R1_001.fastq.gz.bam.gtf,trimmed.57_S147_L003_R1_001.fastq.gz.bam.gtf,trimmed.59_S148_L003_R1_001.fastq.gz.bam.gtf,trimmed.60_S149_L003_R1_001.fastq.gz.bam.gtf,trimmed.61_S150_L003_R1_001.fastq.gz.bam.gtf,trimmed.62_S151_L003_R1_001.fastq.gz.bam.gtf,trimmed.63_S152_L003_R1_001.fastq.gz.bam.gtf,trimmed.64_S153_L003_R1_001.fastq.gz.bam.gtf,trimmed.65_S154_L003_R1_001.fastq.gz.bam.gtf,trimmed.66_S155_L003_R1_001.fastq.gz.bam.gtf,trimmed.67_S156_L003_R1_001.fastq.gz.bam.gtf,trimmed.68_S157_L003_R1_001.fastq.gz.bam.gtf,trimmed.6_S120_L003_R1_001.fastq.gz.bam.gtf
Montipora_capitata_HIv3___RNAseq.g42319.t1,1,0,0,6,3,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,1,0,0,0,0,0,0,2,0,0,0
Montipora_capitata_HIv3___TS.g49315.t1b,42,0,13,115,24,2,130,22,255,191,0,70,113,3,0,6,56,74,306,65,0,178,29,242,9,15,139,3,160,48,114,0,0,27,85,18,88,153,105,116
Montipora_capitata_HIv3___TS.g49315.t1a,325,13,104,1239,669,24,619,56,1441,1052,0,576,825,0,0,19,44,752,1816,1276,0,1018,71,2005,0,79,1047,0,1082,29,1140,0,22,729,1666,96,1287,905,2351,1874
Montipora_capitata_HIv3___RNAseq.g19176.t1,0,0,0,0,0,0,0,0,0,0,0,3,0,0,0,0,0,0,0,0,0,0,0,2,0,0,0,0,0,0,5,0,0,0,0,0,0,0,0,0
Montipora_capitata_HIv3___RNAseq.g42173.t1,0,0,6,6,0,0,6,0,2,3,4,0,2,1,3,0,2,1,2,4,6,0,0,2,2,0,3,2,3,3,4,1,4,0,2,2,6,3,2,0
Montipora_capitata_HIv3___TS.g35745.t1,14,8,10,13,11,19,10,17,17,6,0,7,12,16,36,16,12,28,18,9,33,12,7,38,8,9,9,8,6,3,7,17,8,16,19,17,14,1,8,0
Montipora_capitata_HIv3___RNAseq.g8204.t1,30,45,50,250,14,76,698,778,74,317,72,87,69,337,30,152,3428,45,514,122,136,192,217,595,222,158,290,1409,264,60,555,403,300,378,711,46,103,660,1393,51
Montipora_capitata_HIv3___RNAseq.g9577.t1,14,5,15,10,13,20,11,12,20,3,27,11,12,6,20,9,14,15,4,11,12,9,11,21,13,20,18,21,18,15,23,17,21,6,48,20,22,14,2,15
Montipora_capitata_HIv3___RNAseq.g23056.t1,532,606,1067,1139,601,631,655,661,932,676,511,559,642,774,675,459,446,298,472,307,411,618,1025,801,577,684,764,631,671,943,722,1012,405,362,667,741,1189,611,494,443
Error file:
54384 reference transcripts loaded.
2 duplicate reference transcripts discarded.
54384 query transfrags loaded.
This seems to have fixed the issue with gene naming.
copy gene counts matrix to computer (again)
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/BleachingPairs_RNASeq/KB_gene_count_matrix2.csv /Users/emmastrand/MyProjects/HI_Bleaching_Timeseries/Dec-July-2019-analysis/output/RNASeq/KB_gene_count_matrix2.csv
less merged.stats:
#= Summary for dataset: stringtie_merged.gtf
# Query mRNAs : 54384 in 54185 loci (36023 multi-exon transcripts)
# (141 multi-transcript loci, ~1.0 transcripts per locus)
# Reference mRNAs : 54382 in 54185 loci (36023 multi-exon)
# Super-loci w/ reference transcripts: 54185
#-----------------| Sensitivity | Precision |
Base level: 100.0 | 100.0 |
Exon level: 100.0 | 100.0 |
Intron level: 100.0 | 100.0 |
Intron chain level: 100.0 | 100.0 |
Transcript level: 100.0 | 100.0 |
Locus level: 100.0 | 100.0 |
Matching intron chains: 36023
Matching transcripts: 54361
Matching loci: 54182
Missed exons: 0/256029 ( 0.0%)
Novel exons: 0/256026 ( 0.0%)
Missed introns: 0/201643 ( 0.0%)
Novel introns: 0/201643 ( 0.0%)
Missed loci: 0/54185 ( 0.0%)
Novel loci: 0/54185 ( 0.0%)
Total union super-loci across all input datasets: 54185
54384 out of 54384 consensus transcripts written in merged.annotated.gtf (0 discarded as redundant)
Previous versions of scripts
This took two weeks.. seems long but all worked. Later I found out that the below align.sh actually produced a bam file for both R1 and R2. ah!
align.sh:
#!/bin/bash
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/output/
#SBATCH --error=../"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function (specific need to my computer)
# load modules needed
module load HISAT2/2.2.1-foss-2019b
module load SAMtools/1.9-foss-2018b
# Index the reference genome
hisat2-build -f /data/putnamlab/estrand/Montipora_capitata_HIv3.assembly.fasta ./Mcap_ref
echo "Reference genome indexed. Starting alignment" $(date)
# Alignment of clean reads to the reference genome
array=($(ls /data/putnamlab/estrand/BleachingPairs_RNASeq/processed/*.fastq.gz))
for i in ${array[@]}; do
sample_name=`echo $i| awk -F [.] '{print $2}'`
hisat2 -p 8 --rna-strandness RF --dta -q -x Mcap_ref -1 ${i} -2 $(echo ${i}|sed s/_R1/_R2/) -S ${sample_name}.sam
samtools sort -@ 8 -o ${sample_name}.bam ${sample_name}.sam
echo "${i} bam-ified!"
rm ${sample_name}.sam
done
assemble.sh:
#!/bin/bash
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=128GB
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/output/
#SBATCH --error=../"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function (specific need to my computer)
module load StringTie/2.1.4-GCC-9.3.0
module load gffcompare/0.11.5-foss-2018b
# Assemble and estimate reads
array1=($(ls *.bam))
for i in ${array1[@]}; do
sample_name=`echo $i| awk -F [_] '{print $1"_"$2"_"$3}'`
stringtie -p 8 -e -B -G /data/putnamlab/estrand/Montipora_capitata_HIv3.genes.gff3 -o /data/putnamlab/estrand/BleachingPairs_RNASeq/output/${i}.gtf /data/putnamlab/estrand/BleachingPairs_RNASeq/output/${i}
done
# Merge stringTie gtf results
ls *gtf > mergelist.txt
cat mergelist.txt
stringtie --merge -p 8 -G /data/putnamlab/estrand/Montipora_capitata_HIv3.genes.gff3 -o stringtie_merged.gtf mergelist.txt
echo "Stringtie merge complete" $(date)
genecount.sh:
#!/bin/bash
#SBATCH -t 60:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=128GB
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/BleachingPairs_RNASeq/output/
#SBATCH --error=../"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=../"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function (specific need to my computer)
module load StringTie/2.1.4-GCC-9.3.0
module load Python/2.7.15-foss-2018b
module load GffCompare/0.12.1-GCCcore-8.3.0
# Assess the performance of the assembly
gffcompare -r /data/putnamlab/estrand/Montipora_capitata_HIv3.genes.gff3 -o merged stringtie_merged.gtf
echo "GFFcompare complete, Starting gene count matrix assembly..." $(date)
# Make gtf list text file
for filename in *.bam.gtf; do echo $filename $PWD/$filename; done > listGTF.txt
# Compile the gene count matrix
python ../scripts/prepDE.py -g ../KB_gene_count_matrix.csv -i listGTF.txt
echo "Gene count matrix compiled." $(date)
head output/listGTF.txt :
16_S121_L003_R1_001.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output/16_S121_L003_R1_001.bam.gtf
16_S121_L003_R2_001.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output/16_S121_L003_R2_001.bam.gtf
17_S122_L003_R1_001.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output/17_S122_L003_R1_001.bam.gtf
17_S122_L003_R2_001.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output/17_S122_L003_R2_001.bam.gtf
21_S123_L003_R1_001.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output/21_S123_L003_R1_001.bam.gtf
21_S123_L003_R2_001.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output/21_S123_L003_R2_001.bam.gtf
22_S124_L003_R1_001.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output/22_S124_L003_R1_001.bam.gtf
22_S124_L003_R2_001.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output/22_S124_L003_R2_001.bam.gtf
23_S125_L003_R1_001.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output/23_S125_L003_R1_001.bam.gtf
23_S125_L003_R2_001.bam.gtf /glfs/brick01/gv0/putnamlab/estrand/BleachingPairs_RNASeq/output/23_S125_L003_R2_001.bam.gtf
less less genecount.sh_error.201740:
GCCcore/8.3.0(24):ERROR:150: Module 'GCCcore/8.3.0' conflicts with the currently loaded module(s) 'GCCcore/9.3.0'
GCCcore/8.3.0(24):ERROR:102: Tcl command execution failed: conflict GCCcore
54384 reference transcripts loaded.
2 duplicate reference transcripts discarded.
54384 query transfrags loaded.
head -5 KB_gene_count_matrix.csv:
gene_id,16_S121_L003_R1_001.bam.gtf,16_S121_L003_R2_001.bam.gtf,17_S122_L003_R1_001.bam.gtf,17_S122_L003_R2_001.bam.gtf,21_S123_L003_R1_001.bam.gtf,21_S123_L003_R2_001.bam.gtf,22_S124_L003_R1_001.bam.gtf,22_S124_L003_R2_001.bam.gtf,23_S125_L003_R1_001.bam.gtf,23_S125_L003_R2_001.bam.gtf,24_S126_L003_R1_001.bam.gtf,24_S126_L003_R2_001.bam.gtf,25_S127_L003_R1_001.bam.gtf,25_S127_L003_R2_001.bam.gtf,26_S128_L003_R1_001.bam.gtf,26_S128_L003_R2_001.bam.gtf,28_S129_L003_R1_001.bam.gtf,28_S129_L003_R2_001.bam.gtf,29_S130_L003_R1_001.bam.gtf,29_S130_L003_R2_001.bam.gtf,2_S118_L003_R1_001.bam.gtf,2_S118_L003_R2_001.bam.gtf,30_S131_L003_R1_001.bam.gtf,30_S131_L003_R2_001.bam.gtf,31_S132_L003_R1_001.bam.gtf,31_S132_L003_R2_001.bam.gtf,33_S133_L003_R1_001.bam.gtf,33_S133_L003_R2_001.bam.gtf,37_S134_L003_R1_001.bam.gtf,37_S134_L003_R2_001.bam.gtf,39_S135_L003_R1_001.bam.gtf,39_S135_L003_R2_001.bam.gtf,42_S136_L003_R1_001.bam.gtf,42_S136_L003_R2_001.bam.gtf,43_S137_L003_R1_001.bam.gtf,43_S137_L003_R2_001.bam.gtf,45_S138_L003_R1_001.bam.gtf,45_S138_L003_R2_001.bam.gtf,46_S139_L003_R1_001.bam.gtf,46_S139_L003_R2_001.bam.gtf,47_S140_L003_R1_001.bam.gtf,47_S140_L003_R2_001.bam.gtf,4_S119_L003_R1_001.bam.gtf,4_S119_L003_R2_001.bam.gtf,50_S141_L003_R1_001.bam.gtf,50_S141_L003_R2_001.bam.gtf,51_S142_L003_R1_001.bam.gtf,51_S142_L003_R2_001.bam.gtf,52_S143_L003_R1_001.bam.gtf,52_S143_L003_R2_001.bam.gtf,54_S144_L003_R1_001.bam.gtf,54_S144_L003_R2_001.bam.gtf,55_S145_L003_R1_001.bam.gtf,55_S145_L003_R2_001.bam.gtf,56_S146_L003_R1_001.bam.gtf,56_S146_L003_R2_001.bam.gtf,57_S147_L003_R1_001.bam.gtf,57_S147_L003_R2_001.bam.gtf,59_S148_L003_R1_001.bam.gtf,59_S148_L003_R2_001.bam.gtf,60_S149_L003_R1_001.bam.gtf,60_S149_L003_R2_001.bam.gtf,61_S150_L003_R1_001.bam.gtf,61_S150_L003_R2_001.bam.gtf,62_S151_L003_R1_001.bam.gtf,62_S151_L003_R2_001.bam.gtf,63_S152_L003_R1_001.bam.gtf,63_S152_L003_R2_001.bam.gtf,64_S153_L003_R1_001.bam.gtf,64_S153_L003_R2_001.bam.gtf,65_S154_L003_R1_001.bam.gtf,65_S154_L003_R2_001.bam.gtf,66_S155_L003_R1_001.bam.gtf,66_S155_L003_R2_001.bam.gtf,67_S156_L003_R1_001.bam.gtf,67_S156_L003_R2_001.bam.gtf,68_S157_L003_R1_001.bam.gtf,68_S157_L003_R2_001.bam.gtf,6_S120_L003_R1_001.bam.gtf,6_S120_L003_R2_001.bam.gtf
Montipora_capitata_HIv3___RNAseq.g19176.t1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,3,0,0,0,0,2,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,2,0,0,0,0,0,0,0,0,5,0,0,0,0,5,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0
Montipora_capitata_HIv3___RNAseq.g42173.t1,0,0,0,6,6,11,6,13,0,3,0,1,6,0,0,10,2,3,3,7,4,82,0,0,2,1,1,0,3,3,0,6,2,5,1,6,2,5,4,4,6,2,0,4,0,7,2,0,2,0,0,3,3,5,2,5,3,5,3,7,4,4,1,0,4,52,0,5,2,4,2,0,6,8,3,11,2,7,0,4
Montipora_capitata_HIv3___RNAseq.g13553.t1,0,0,0,0,0,0,1,1,0,1,0,0,0,1,0,0,0,0,0,0,0,0,0,0,0,1,0,0,0,2,0,0,0,0,0,0,10,6,0,0,0,0,0,0,0,0,0,0,0,0,0,0,11,6,0,0,0,0,0,0,0,0,0,2,0,2,0,0,1,2,0,0,0,0,0,0,0,0,0,1
STRG.6173,396,227,1108,635,799,461,924,535,554,312,1959,1154,931,549,2016,1178,1126,663,1014,604,874,516,1169,689,1574,931,1471,867,1542,912,1525,908,1370,815,1739,1011,716,428,826,488,716,415,1109,651,866,519,2429,1434,1124,664,804,487,1575,957,1524,936,1585,934,822,488,1219,720,1646,973,1169,716,1282,764,1598,972,1915,1165,1465,877,1331,799,915,556,1016,590
Column with gene ID contains both gene names from reference genome and IDs from this pipeline (e.g. STRG.6173). Those from this pipeline seem to have the highest counts.. What does this mean? see troubleshooting section below
Copy that output to local computer
``` scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/BleachingPairs_RNASeq/KB_gene_count_matrix.csv /Users/emmastrand/MyProjects/HI_Bleaching_Timeseries/Dec-July-2019-analysis/output/RNASeq/KB_gene_count_matrix.csv