
E5 16S Analysis
16S Analysis for Mo’orea E5 project
12 test samples from the Mo’orea E5 project. Used my pipeline from Holobiont Integration project (found here).
Primers used:
- 515F: GTGYCAGCMGCCGCGGTAA
- 806RB: GGACTACNVGGGTWTCTAAT
Modules needed:
- FastQC/0.11.9-Java-11
- MultiQC/1.9-intel-2020a-Python-3.8.2
- QIIME2/2021.4
Secure copy paste 12 files to bluewaves server
Ran outside of bluewaves / before logging into bluewaves and andromeda.
$ scp -r /Users/emmastrand/MyProjects/EmmaStrand_Notebook/16S_E5/raw_files emma_strand@bluewaves.uri.edu:/data/putnamlab/estrand/16S_E5
FastQC
Build and run fastqc script
Script: putnamlab/estrand/16S_E5/scripts/fastqc.sh
# within 16S_E5 folder
$ mkdir fastqc_results
Ran below script.
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=emma_strand@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/16S_E5/raw_files
#SBATCH --error="script_error" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
for file in /data/putnamlab/estrand/16S_E5/raw_files/*fastq.gz
do
fastqc $file --outdir /data/putnamlab/estrand/16S_E5/fastqc_results
done
multiqc --interactive /data/putnamlab/estrand/16S_E5/fastqc_results
mv multiqc_report.html 16S_raw_qc_multiqc_report_E5.html #renames file
Double check all files were processed. Output should be 12 x 2 (F and R reads) x 2 (html and zip file) = 48.
$ cd /data/putnamlab/estrand/16S_E5/fastqc_results
$ ls -1 | wc -l
output:
48
Copy and paste to notebook (outside of andromeda in a new terminal tab)
$ scp emma_strand@bluewaves.uri.edu:/data/putnamlab/estrand/16S_E5/raw_files/16S_raw_qc_multiqc_report_E5.html /Users/emmastrand/MyProjects/EmmaStrand_Notebook/16S_E5/
Link to full report: Emma Strand Github Notebook
Raw QC Results
Download the full multiqc report from the link above to view all aspects of quality assessment.
Check duplicated reads and per sequence GC content specifically from this report.
Mean Quality Scores: Red = R1 (forward). Blue = R2 (reverse)
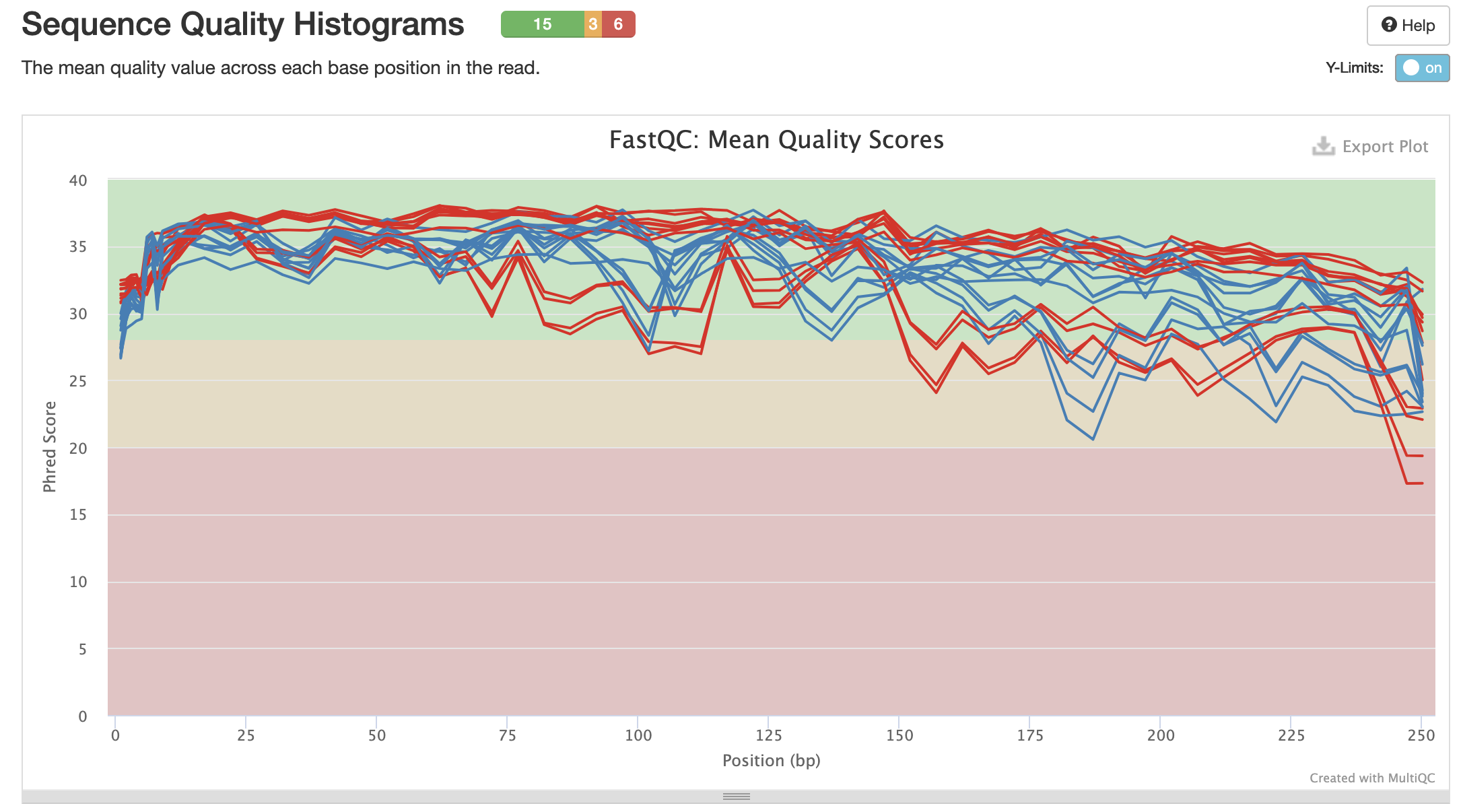
The quality scores will be what I based the trimming and truncating steps in the QIIME2 pipeline off of.
Moving files out of raw_files folder.
mv 16S_raw_qc_multiqc_report_E5.html ../16S_raw_qc_multiqc_report_E5.html
mv multiqc_data ../multiqc_data
mv output_script ../output_script
mv script_error ../script_error
QIIME2
Create metadata directory
$ cd /data/putnamlab/estrand/16S_E5
$ mkdir metadata
Creating metadata files from list of raw_files and in R
R Script link: Emma Strand Notebook link
Create list of file names in andromeda.
$ cd 16S_E5
$ find raw_files -type f -print | sed 's_/_,_g' > ~/filelist.csv
$ mv ~/filelist.csv /data/putnamlab/estrand/16S_E5
Move that file to desktop to be able to run in R.
scp emma_strand@bluewaves.uri.edu:/data/putnamlab/estrand/16S_E5/filelist.csv /Users/emmastrand/MyProjects/EmmaStrand_Notebook/16S_E5/
1. Sample manifest file
Created in R (csv file) and scp to bluewaves folder
scp /Users/emmastrand/MyProjects/EmmaStrand_Notebook/16S_E5/sample_manifest_16S_E5.csv emma_strand@bluewaves.uri.edu:/data/putnamlab/estrand/16S_E5/metadata
2. Sample metadata file
Created in R (txt file) and scp to bluewaves folder
scp /Users/emmastrand/MyProjects/EmmaStrand_Notebook/16S_E5/metadata.txt emma_strand@bluewaves.uri.edu:/data/putnamlab/estrand/16S_E5/metadata
Sample Import
$ cd scripts
$ nano sample_import.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=emma_strand@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/16S_E5/raw_files
#SBATCH --error="script_error" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
module load QIIME2/2021.4
cd /data/putnamlab/estrand/16S_E5/raw_files
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \
--input-path /data/putnamlab/estrand/16S_E5/metadata/sample_manifest_16S_E5.csv \
--input-format PairedEndFastqManifestPhred33 \
--output-path ../processed_data/E5_16S-paired-end-sequences.qza
Denoising with DADA2
$ cd scripts
$ nano denoise.sh
P-trim values are based on primer length:
- p-trim-left-r 21 (reverse is 21 bp long)
- p-trim-left-f 19 (forward is 19 bp long)
P-truncate values are based on where the mean quality scores of R1 and R2 files start to decrease:
- p-trunc-len-r 175
- p-trunc-len-f 150
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=emma_strand@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/16S_E5/raw_files
#SBATCH --error="script_error" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
module load QIIME2/2021.4
cd /data/putnamlab/estrand/16S_E5/processed_data
# Metadata path
METADATA="../metadata/metadata.txt"
qiime dada2 denoise-paired --verbose --i-demultiplexed-seqs E5_16S-paired-end-sequences.qza \
--p-trunc-len-r 175 --p-trunc-len-f 150 \
--p-trim-left-r 19 --p-trim-left-f 21 \
--o-table table.qza \
--o-representative-sequences rep-seqs.qza \
--o-denoising-stats denoising-stats.qza \
--p-n-threads 20
# Summarize feature table and sequences
qiime metadata tabulate \
--m-input-file denoising-stats.qza \
--o-visualization denoising-stats.qzv
qiime feature-table summarize \
--i-table table.qza \
--o-visualization table.qzv \
--m-sample-metadata-file $METADATA
qiime feature-table tabulate-seqs \
--i-data rep-seqs.qza \
--o-visualization rep-seqs.qzv
Results:
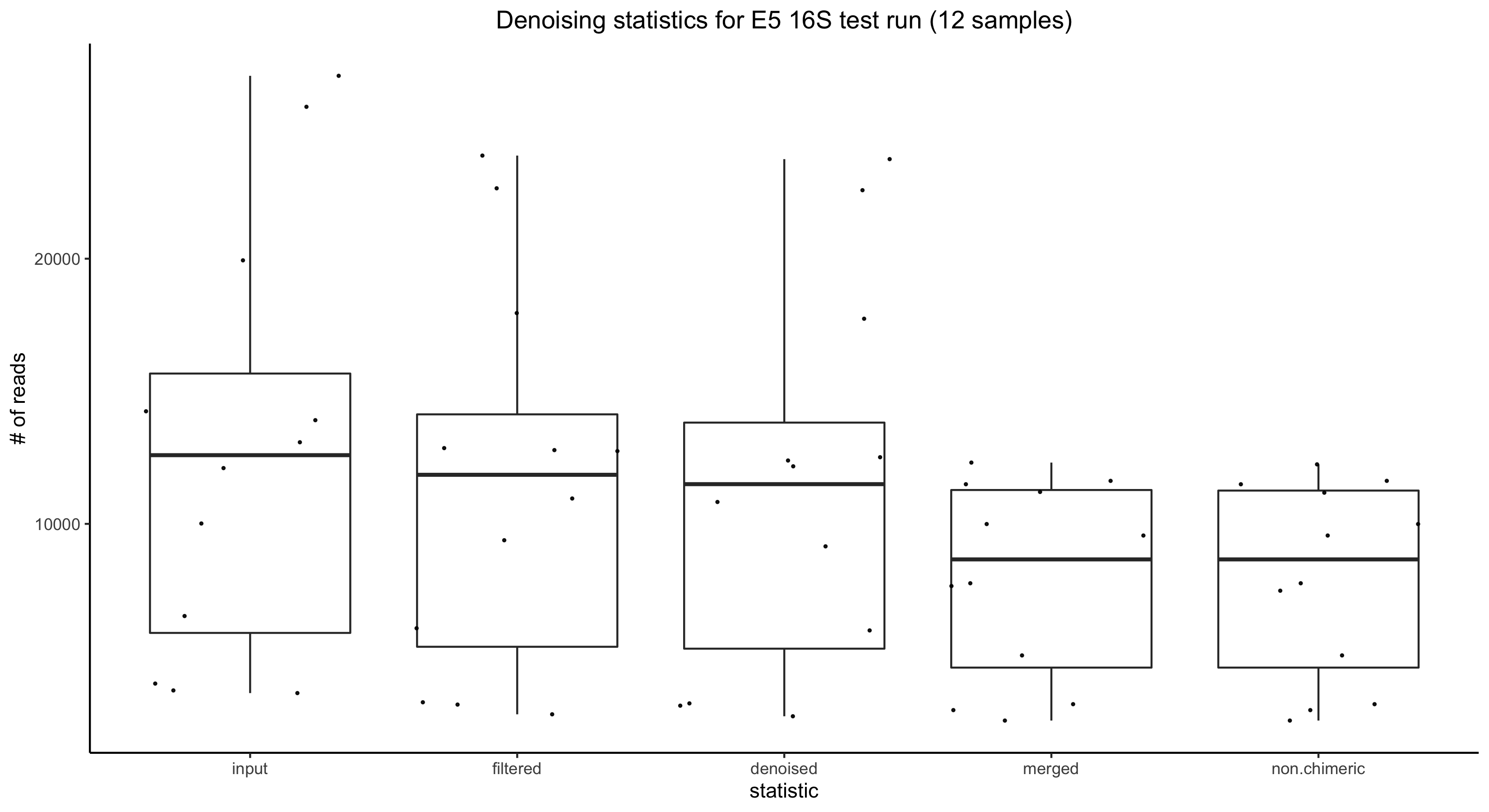
Taxonomic Identification
Classifier: classify-sklearn. This was recommended by QIIME2.
database: silva-138-99-515-806-nb-classifier.qza. This was most relevant to the primers used.
Download the classifier database
$ cd metadata
$ wget https://data.qiime2.org/2021.4/common/silva-138-99-515-806-nb-classifier.qza
Unfiltered version
$ cd scripts
$ nano taxonomic_id.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=emma_strand@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/16S_E5/raw_files
#SBATCH --error="script_error" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
module load QIIME2/2021.4
cd /data/putnamlab/estrand/16S_E5/processed_data
# Metadata path
METADATA="../metadata/metadata.txt"
qiime feature-classifier classify-sklearn \
--i-classifier ../metadata/silva-138-99-515-806-nb-classifier.qza \
--i-reads rep-seqs.qza \
--o-classification taxonomy.qza
qiime metadata tabulate \
--m-input-file taxonomy.qza \
--o-visualization taxonomy.qzv
qiime taxa barplot \
--i-table table.qza \
--i-taxonomy taxonomy.qza \
--m-metadata-file $METADATA \
--o-visualization taxa-bar-plots.qzv
qiime metadata tabulate \
--m-input-file rep-seqs.qza \
--m-input-file taxonomy.qza \
--o-visualization tabulated-feature-metadata.qzv
Filtered version
This filters the above tables generated. You need to run the above taxonomic_id.sh script prior to running taxonomic_id_filtered.sh.
$ cd scripts
$ nano taxonomic_id_filtered.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=emma_strand@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/estrand/16S_E5/raw_files
#SBATCH --error="script_error" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
module load QIIME2/2021.4
cd /data/putnamlab/estrand/16S_E5/processed_data
# Metadata path
METADATA="../metadata/metadata.txt"
qiime taxa filter-table \
--i-table table.qza \
--i-taxonomy taxonomy.qza \
--p-mode contains \
--p-exclude "Unassigned","Chloroplast","Eukaryota" \
--o-filtered-table table-filtered.qza
qiime metadata tabulate \
--m-input-file taxonomy.qza \
--o-visualization taxonomy.qzv
qiime taxa barplot \
--i-table table-filtered.qza \
--i-taxonomy taxonomy.qza \
--m-metadata-file $METADATA \
--o-visualization taxa-bar-plots-filtered.qzv
qiime metadata tabulate \
--m-input-file rep-seqs.qza \
--m-input-file taxonomy.qza \
--o-visualization tabulated-feature-metadata.qzv
Move files of interest to desktop/github
View the below files in QIIME2 view.
## Denoising stats
scp -r emma_strand@bluewaves.uri.edu:/data/putnamlab/estrand/16S_E5/processed_data/rep-seqs.qzv /Users/emmastrand/MyProjects/EmmaStrand_Notebook/16S_E5/rep-seqs.qzv
## Rep Seqs
scp -r emma_strand@bluewaves.uri.edu:/data/putnamlab/estrand/16S_E5/processed_data/denoising-stats.qzv /Users/emmastrand/MyProjects/EmmaStrand_Notebook/16S_E5/denoising-stats.qzv
## Taxa Bar Plots
# Unfiltered taxa bar plots
scp -r emma_strand@bluewaves.uri.edu:/data/putnamlab/estrand/16S_E5/processed_data/taxa-bar-plots.qzv /Users/emmastrand/MyProjects/EmmaStrand_Notebook/16S_E5/taxa-bar-plots.qzv
# Filtered taxa bar plots
scp -r emma_strand@bluewaves.uri.edu:/data/putnamlab/estrand/16S_E5/processed_data/taxa-bar-plots-filtered.qzv /Users/emmastrand/MyProjects/EmmaStrand_Notebook/16S_E5/taxa-bar-plots-filtered.qzv
Download and input in qiime2 view: