
16S-V3V4 Test Run Analysis Pipeline
Analysis Pipeline for 16S V3V4 Test Sequencing Run 1
Lab work that produced these sequences: https://github.com/emmastrand/EmmaStrand_Notebook/blob/master/_posts/2022-03-21-16S-V3V4-Sample-Processing.md
Questions to answer:
- Do we get microbiome or host sequences from 338F and 341F V3V4 region PCR products?
- Which V3V34 primer (338F and or 341F) yields higher microbiome sequences?
- Does 63C annealing temperature for the V4 (515F) region yield better microbiome vs host ratios? Is this still all host?
- What kind of noise do we get from negative controls?
Contents:
- Setting Up Andromeda
- Initial fastqc on files
- Initial Multiqc Report
- Separate 16S projects
- Create metadata files
- QIIME2: Sample Import
- QIIME2: Denoise
- QIIME2: Taxonomic ID
Test results with other 16S runs in this google sheet: https://docs.google.com/spreadsheets/d/1ZHO469WzDxJ7PwNkERvx11j54PQk54sZM_B1KnaKIt0/edit#gid=1005240003
Setting Up Andromeda
Make a new directory for the test output, fastqc results, and scripts.
$ mkdir /data/putnamlab/estrand/Test_V3V4_16S
$ cd Test_V3V4_16S
$ mkdir scripts
$ mkdir fastqc_results
Raw data path: /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral. Contains md5 checksum files, download from basespace script (.sh), and ELS01-30 R1 and R2 files (60 total).
Initial fastqc on files
fastqc.sh created in the scripts folder. This takes under 10 minutes.
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=emma_strand@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --error="script_error_fastqc" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_fastqc" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
cd /data/putnamlab/estrand/Test_V3V4_16S
for file in /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/*fastq.gz
do
fastqc $file --outdir /data/putnamlab/estrand/Test_V3V4_16S/fastqc_results
done
multiqc --interactive /data/putnamlab/estrand/Test_V3V4_16S/fastqc_results ### this file will output in whatever directory we cd'd to first
mv multiqc_report.html V3V4Test_initial_multiqc_report.html # renames file
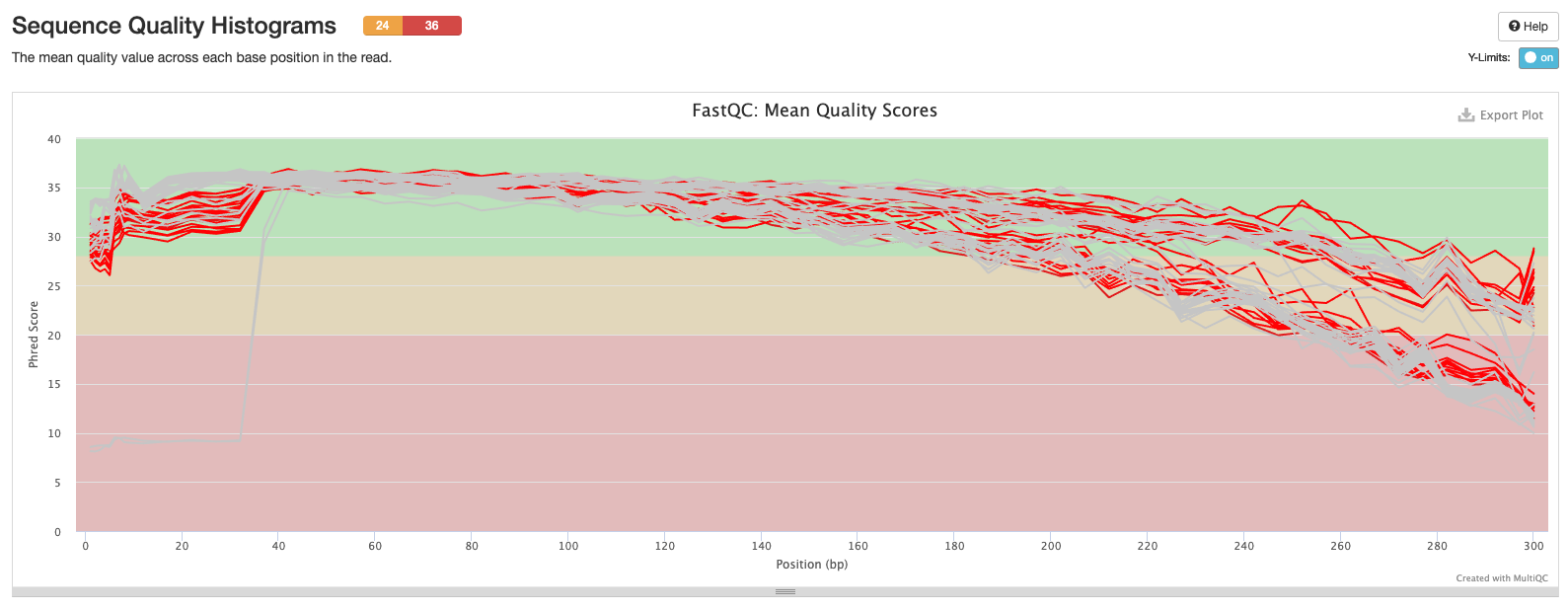
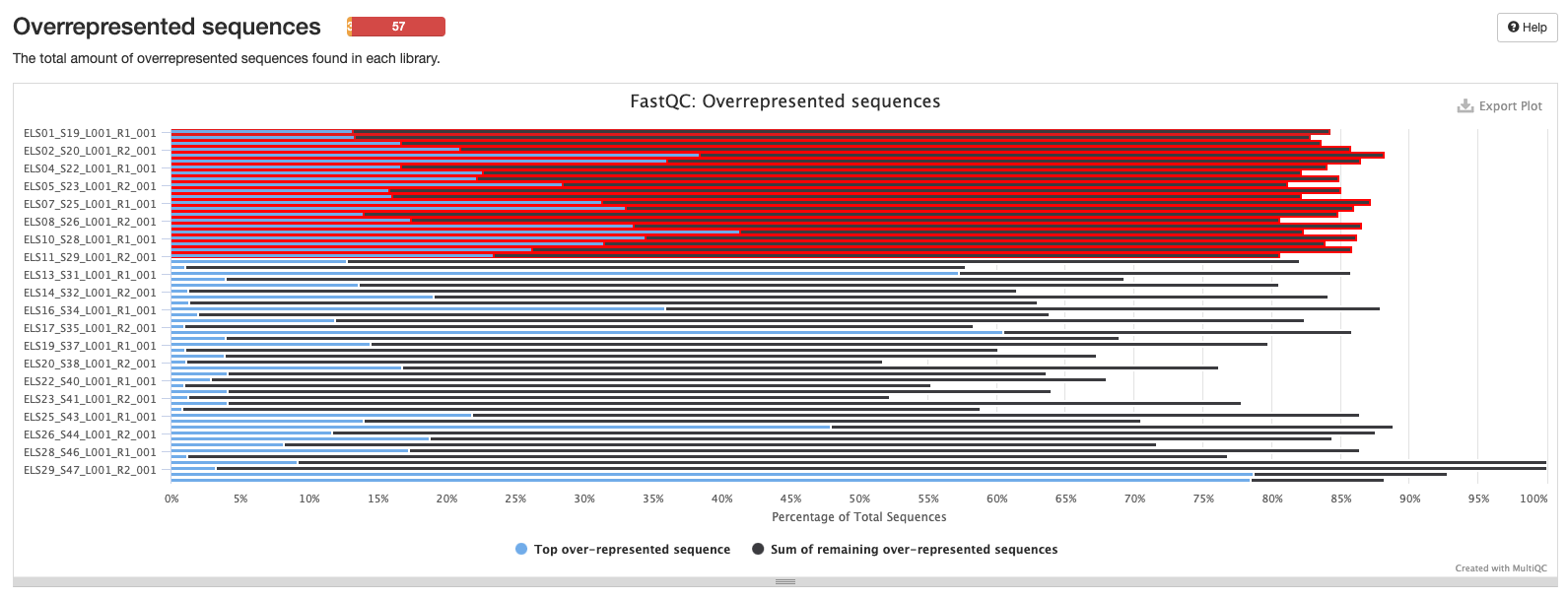
Initial MultiQC Report
All samples run together. The RED indicates ITS2 samples, the grey indicates 16S samples.
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/V3V4Test_initial_multiqc_report.html /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4Test_initial_multiqc_report.html
Full multiqc report here: https://github.com/emmastrand/EmmaStrand_Notebook/blob/master/Lab-work/V3V4Test_initial_multiqc_report.html







Separate 16S projects
Made a new directory sample_sets. 1.) V3V4_338F. 2.) V3V4_341F. 3.) V4_515F.
Copying files into each folder:
1.) V3V4_338F
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS12_S30_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_338F
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS13_S31_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_338F
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS14_S32_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_338F
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS15_S33_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_338F
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS16_S34_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_338F
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS17_S35_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_338F
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS18_S36_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_338F
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS19_S37_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_338F
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS28_S46_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_338F
2.) V3V4_341F
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS20_S38_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_341F
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS21_S39_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_341F
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS22_S40_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_341F
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS23_S41_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_341F
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS24_S42_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_341F
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS29_S47_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_341F
3.) V4_515F.
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS25_S43_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V4_515F
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS26_S44_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V4_515F
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS27_S45_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V4_515F
$ cp /data/putnamlab/KITT/hputnam/2022728_16sTest_Coral/ELS30_S48_L001_R{1,2}_001.fastq.gz /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V4_515F
Cut-offs for each set of primers:
Based on sequence quality
- Truncating R = 220
- Truncating F = 250
Based on primer length
- Trim R = 20 (all 806R)
- Trim F = 19 (338F and 515F)
- Trim F = 17 (341F)
Create metadata files
1. Sample manifest files
$ cd V3V4_338F
$ find raw_data -type f -print | sed 's_/_,_g' > metadata/filelist_V3V4_338F.csv
$ cd V3V4_341F
$ find raw_data -type f -print | sed 's_/_,_g' > metadata/filelist_V3V4_341F.csv
$ cd V4_515F
$ find raw_data -type f -print | sed 's_/_,_g' > metadata/filelist_V4_515F.csv
copy those outside of andromeda
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_338F/metadata/filelist_V3V4_338F.csv /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_341F/metadata/filelist_V3V4_341F.csv /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V4_515F/metadata/filelist_V4_515F.csv /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/
Run R metadata creation script and scp back onto andromeda (in a window outside andromeda)
scp /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/sample_manifest338.txt emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_338F/metadata/sample_manifest338.txt
scp /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/sample_manifest341.txt emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_341F/metadata/sample_manifest341.txt
scp /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/sample_manifest515.txt emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V4_515F/metadata/sample_manifest515.txt
2. Sample metadata list
Run R metadata creation script and scp back onto andromeda (in a window outside andromeda)
scp /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/metadata338.txt emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_338F/metadata/metadata338.txt
scp /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/metadata341.txt emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_341F/metadata/metadata341.txt
scp /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/metadata515.txt emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V4_515F/metadata/metadata515.txt
QIIME2: Sample Import
338F
This took about 2 minutes.
import338.sh:
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=emma_strand@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --error="script_error_import338" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_import338" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
module load QIIME2/2021.8
#### METADATA FILES ####
# File path -- change this to correspond to what script you are running
cd /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_338F/
# Metadata path
METADATA="metadata/metadata338.txt"
# Sample manifest path
MANIFEST="metadata/sample_manifest338.txt"
#########################
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \
--input-path $MANIFEST \
--input-format PairedEndFastqManifestPhred33V2 \
--output-path V3V4_338-paired-end-sequences.qza
341F
import341.sh:
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=emma_strand@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --error="script_error_import341" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_import341" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
module load QIIME2/2021.8
#### METADATA FILES ####
# File path -- change this to correspond to what script you are running
cd /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_341F/
# Metadata path
METADATA="metadata/metadata341.txt"
# Sample manifest path
MANIFEST="metadata/sample_manifest341.txt"
#########################
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \
--input-path $MANIFEST \
--input-format PairedEndFastqManifestPhred33V2 \
--output-path V3V4_341-paired-end-sequences.qza
515F
import515.sh:
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=emma_strand@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --error="script_error_import515" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_import515" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
module load QIIME2/2021.8
#### METADATA FILES ####
# File path -- change this to correspond to what script you are running
cd /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V4_515F/
# Metadata path
METADATA="metadata/metadata515.txt"
# Sample manifest path
MANIFEST="metadata/sample_manifest515.txt"
#########################
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \
--input-path $MANIFEST \
--input-format PairedEndFastqManifestPhred33V2 \
--output-path V3V4_515-paired-end-sequences.qza
QIIME2: Denoise
338F
See multiqc section for parameter choices
denoise338.sh:
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=emma_strand@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --error="script_error_denoise338" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_denoise338" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
module load QIIME2/2021.8
#### METADATA FILES ####
# File path -- change this to correspond to what script you are running
cd /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_338F/
# Metadata path
METADATA="metadata/metadata338.txt"
# Sample manifest path
MANIFEST="metadata/sample_manifest338.txt"
#########################
qiime dada2 denoise-paired --verbose --i-demultiplexed-seqs V3V4_338-paired-end-sequences.qza \
--p-trunc-len-r 220 --p-trunc-len-f 250 \
--p-trim-left-r 20 --p-trim-left-f 19 \
--o-table table_338.qza \
--o-representative-sequences rep-seqs_338.qza \
--o-denoising-stats denoising-stats_338.qza \
--p-n-threads 20
#### CLUSTERING
# Summarize feature table and sequences
qiime metadata tabulate \
--m-input-file denoising-stats_338.qza \
--o-visualization denoising-stats_338.qzv
qiime feature-table summarize \
--i-table table_338.qza \
--o-visualization table_338.qzv \
--m-sample-metadata-file $METADATA
qiime feature-table tabulate-seqs \
--i-data rep-seqs_338.qza \
--o-visualization rep-seqs_338.qzv
341F
See multiqc section for parameter choices
denoise341.sh:
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=emma_strand@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --error="script_error_denoise341" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_denoise341" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
module load QIIME2/2021.8
#### METADATA FILES ####
# File path -- change this to correspond to what script you are running
cd /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_341F/
# Metadata path
METADATA="metadata/metadata341.txt"
# Sample manifest path
MANIFEST="metadata/sample_manifest341.txt"
#########################
qiime dada2 denoise-paired --verbose --i-demultiplexed-seqs V3V4_341-paired-end-sequences.qza \
--p-trunc-len-r 220 --p-trunc-len-f 250 \
--p-trim-left-r 20 --p-trim-left-f 17 \
--o-table table_341.qza \
--o-representative-sequences rep-seqs_341.qza \
--o-denoising-stats denoising-stats_341.qza \
--p-n-threads 20
#### CLUSTERING
# Summarize feature table and sequences
qiime metadata tabulate \
--m-input-file denoising-stats_341.qza \
--o-visualization denoising-stats_341.qzv
qiime feature-table summarize \
--i-table table_341.qza \
--o-visualization table_341.qzv \
--m-sample-metadata-file $METADATA
qiime feature-table tabulate-seqs \
--i-data rep-seqs_341.qza \
--o-visualization rep-seqs_341.qzv
515F
See multiqc section for parameter choices
denoise515.sh:
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=emma_strand@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --error="script_error_denoise515" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_denoise515" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
module load QIIME2/2021.8
#### METADATA FILES ####
# File path -- change this to correspond to what script you are running
cd /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V4_515F/
# Metadata path
METADATA="metadata/metadata515.txt"
# Sample manifest path
MANIFEST="metadata/sample_manifest515.txt"
#########################
qiime dada2 denoise-paired --verbose --i-demultiplexed-seqs V3V4_515-paired-end-sequences.qza \
--p-trunc-len-r 220 --p-trunc-len-f 250 \
--p-trim-left-r 20 --p-trim-left-f 19 \
--o-table table_515.qza \
--o-representative-sequences rep-seqs_515.qza \
--o-denoising-stats denoising-stats_515.qza \
--p-n-threads 20
#### CLUSTERING
# Summarize feature table and sequences
qiime metadata tabulate \
--m-input-file denoising-stats_515.qza \
--o-visualization denoising-stats_515.qzv
qiime feature-table summarize \
--i-table table_515.qza \
--o-visualization table_515.qzv \
--m-sample-metadata-file $METADATA
qiime feature-table tabulate-seqs \
--i-data rep-seqs_515.qza \
--o-visualization rep-seqs_515.qzv
copying outside terminal to work with in R
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_338F/denoising-stats_338.qzv /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_341F/denoising-stats_341.qzv /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V4_515F/denoising-stats_515.qzv /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/
Open qiime2 view and drop in the first file you want to view. Click ‘Download metadata TSV file’ and save that file to ‘~/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/’ folder.
Run the denoising stats portion of the ‘16S_metadata.R’ script and then return to the following steps below.


This isn’t super encouraging as 20% is much lower than our other projects output is..
For reference, see our results from V4 with oyster microbiome: https://github.com/emmastrand/EmmaStrand_Notebook/blob/master/_posts/2022-03-07-Point-Judith-Oyster-Gut-16S-V6-Analysis.md#denoise-paramter-trials and Kevin’s Porites was ~45% kept by the end of denoising statistics.
QIIME2: Taxonomic ID
For 338F and 341F use full database for now and make a V3V4 specific on later on if we do full analysis:
$ cd /data/putnamlab/estrand/Test_V3V4_16S
$ wget https://data.qiime2.org/2021.11/common/silva-138-99-nb-classifier.qza
# name of file: silva-138-99-nb-classifier.qza
For 515F use the silva databse I used for HI project: /data/putnamlab/estrand/BleachingPairs_16S/metadata/silva-138-99-515-806-nb-classifier.qza
338F
tax338.sh:
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=emma_strand@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --error="script_error_taxonomy338" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_taxonomy338" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
module load QIIME2/2021.8
#### METADATA FILES ####
# File path -- change this to correspond to what script you are running
cd /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_338F
# Metadata path
METADATA="metadata/metadata338.txt"
# Sample manifest path
MANIFEST="metadata/sample_manifest338.txt"
#########################
#### TAXONOMY CLASSIFICATION
qiime feature-classifier classify-sklearn \
--i-classifier /data/putnamlab/estrand/Test_V3V4_16S/silva-138-99-nb-classifier.qza \
--i-reads rep-seqs_338.qza \
--o-classification taxonomy_338.qza
## UNFILTERED
qiime metadata tabulate \
--m-input-file taxonomy_338.qza \
--o-visualization taxonomy_338.qzv
qiime taxa barplot \
--i-table table_338.qza \
--i-taxonomy taxonomy_338.qza \
--m-metadata-file $METADATA \
--o-visualization taxa-bar-plots338.qzv
qiime metadata tabulate \
--m-input-file rep-seqs_338.qza \
--m-input-file taxonomy_338.qza \
--o-visualization tabulated-feature-metadata338.qzv
341F
tax341.sh:
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=emma_strand@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --error="script_error_taxonomy341" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_taxonomy341" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
module load QIIME2/2021.8
#### METADATA FILES ####
# File path -- change this to correspond to what script you are running
cd /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_341F
# Metadata path
METADATA="metadata/metadata341.txt"
# Sample manifest path
MANIFEST="metadata/sample_manifest341.txt"
#########################
#### TAXONOMY CLASSIFICATION
qiime feature-classifier classify-sklearn \
--i-classifier /data/putnamlab/estrand/Test_V3V4_16S/silva-138-99-nb-classifier.qza \
--i-reads rep-seqs_341.qza \
--o-classification taxonomy_341.qza
## UNFILTERED
qiime metadata tabulate \
--m-input-file taxonomy_341.qza \
--o-visualization taxonomy_341.qzv
qiime taxa barplot \
--i-table table_341.qza \
--i-taxonomy taxonomy_341.qza \
--m-metadata-file $METADATA \
--o-visualization taxa-bar-plots341.qzv
qiime metadata tabulate \
--m-input-file rep-seqs_341.qza \
--m-input-file taxonomy_341.qza \
--o-visualization tabulated-feature-metadata341.qzv
515F
tax515.sh:
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=emma_strand@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --error="script_error_taxonomy515" #if your job fails, the error report will be put in this file
#SBATCH --output="output_script_taxonomy515" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
module load QIIME2/2021.8
#### METADATA FILES ####
# File path -- change this to correspond to what script you are running
cd /data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V4_515F
# Metadata path
METADATA="metadata/metadata515.txt"
# Sample manifest path
MANIFEST="metadata/sample_manifest515.txt"
#########################
#### TAXONOMY CLASSIFICATION
qiime feature-classifier classify-sklearn \
--i-classifier /data/putnamlab/estrand/BleachingPairs_16S/metadata/silva-138-99-515-806-nb-classifier.qza \
--i-reads rep-seqs_515.qza \
--o-classification taxonomy_515.qza
## UNFILTERED
qiime metadata tabulate \
--m-input-file taxonomy_515.qza \
--o-visualization taxonomy_515.qzv
qiime taxa barplot \
--i-table table_515.qza \
--i-taxonomy taxonomy_515.qza \
--m-metadata-file $METADATA \
--o-visualization taxa-bar-plots515.qzv
qiime metadata tabulate \
--m-input-file rep-seqs_515.qza \
--m-input-file taxonomy_515.qza \
--o-visualization tabulated-feature-metadata515.qzv
copying output to desktop outside of andromeda
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_338F/taxa-bar-plots338.qzv /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_338F/table_338.qzv /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_341F/taxa-bar-plots341.qzv /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V3V4_341F/table_341.qzv /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V4_515F/taxa-bar-plots515.qzv /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/
scp emma_strand@ssh3.hac.uri.edu:../../data/putnamlab/estrand/Test_V3V4_16S/sample_sets/V4_515F/table_515.qzv /Users/emmastrand/MyProjects/EmmaStrand_Notebook/Lab-work/V3V4_test/
Table summary results (unfiltered)

338F Taxonomic Results (unfiltered)

341F Taxonomic Results (unfiltered)

515F Taxonomic Results (unfiltered)
 )
)
Results summary
I cannot get Mcapitata adult fragments to show a band on the gel post PCR with either 338F or 341F primers, which is why there are only settled recruits and eggs for M. capitata in the 338 and 341 sections.
There are a ton of PCR inhibitors in tissue homogenate - we could dilute the input to try to yield higher microbioal sequences and use a DNA only kit that is made to target microbioal sequences. But the nature of us going DNA/RNA duet extractions is high priority for gene exp and DNA methylation from the same genomic input. Our 16S work may be better off as separate extractions.
1.) Do we get microbiome or host sequences from 338F and 341F V3V4 region PCR products?
We get a majority of sequences to be Cyanobacteria and Proteobacteria with both 338F and 341F primers. We don’t get the levels of high general D:Bacteria sequences that we do with 515F which is great! But I’m concerned with the low % and # read values at the end of QIIME2 denoising section. 2,000 read depth is passable but 5-10k is a better range to shoot for.
2.) Which V3V34 primer (338F and or 341F) yields higher microbiome sequences?
There doesn’t appear to be a substantial visual difference between 338F and 341F. See KW10 Porites, Mcap Plug 10 settled recruit, Mcap D1 eggs, and Pacuta 2513 adult samples for comparison. These appear to be very similar results which is relieving and gives me more confidence in our methods. However, I still cannot get adult Mcapitata fragments to work with either 338F or 341F (no band shows up on gel).
3.) Does 63C annealing temperature for the V4 (515F) region yield better microbiome vs host ratios? Is this still all host?
This seems to be variable with one adult Mcapitata fragment yielding high general bacteria and one with little to no general bacteria. I don’t think this is an annealing temperature issue rather than a V4 only region specific issue.
4.) What kind of noise do we get from negative controls?
This seems to really variable.. The 338F negative control had many reads that passed filtering and were assigned to taxonomic units while the negative control from 341F and 515F did not.
338F: This negative control has the same # of reads as the samples which is not what we want and more less renders the samples un-usable. This could be in well to well contamination. We can look at the composition of neighboring wells and compare to the composition of the negative control.
341F: Lower # of readss than the samples and no bacteria reads assigned, yay! Great.
515F: This still has lower # of reads, not zero but lower than 5,000 sub samples threshold which is great.
Alternatives: Do an extraction negative control and sequence multiple negative controls. If there is high # of reads, view to a more in depth tax unit and see if these are human contamination (i.e. someone breathing on it but with masks that shouldn’t be an issue).
Final thoughts
Especially because I cannot get Mcapitata fragments to show up with either of the V3V4 primers, I’m not super confident this is worth pursuing.. Maybe Pacuta only for Holobiont Integration but not KBay adult Mcapitata samples.
Wesley is going to reach out to Austin from Donahue lab b/c he successfully sequenced Mcap microbiome too. Nelson’s lab hasn’t done extractions in a couple years and are going to try to sequence KBay project fully for 16S so if theirs works then we don’t need to pursue KBay Mcap samples at URI.